

基于Nature Communications文献(2026) | DOI: 10.1038/s41467-026-70947-6
Copper-hydroxyl interactions drive water-promoted copper surface oxidation and mobility
关键词:反应力场 | 铜表面氧化 | 分子动力学 | 水促进 | 界面机理
一、论文简介
氧气(O₂)和水(H₂O)在金属表面的反应是异相催化、腐蚀和电化学能量转换等领域的关键过程。然而,由于O₂和H₂O均产生相同的解离产物——原子氧(O),并且共享羟基(OH)中间体,长期以来难以厘清两者在金属氧化中的各自作用。
Li等人于2026年在Nature Communications发表了题为“Copper-hydroxyl interactions drive water-promoted copper surface oxidation and mobility”的研究论文。该工作结合原位透射电子显微镜(in-situ TEM)技术和ReaxFF反应力场分子动力学模拟,系统揭示了水分子在铜表面氧化中的促进作用机制。
—— 核心发现:Cu/CuOₓ界面上Cu–OH强相互作用引发界面结构动态无序,降低氧的嵌入阻力,加速铜表面氧化进程 ——
二、仿真步骤
2.1 ReaxFF反应力场选择
本研究采用ReaxFF反应力场描述Cu–O–H体系中的化学反应过程。ReaxFF力场通过键级(Bond Order)概念动态处理化学键的断裂与形成,能够准确描述金属氧化过程中涉及的Cu–O键断裂、O–H键解离以及Cu–OH键形成等反应事件。
力场参数经过密度泛函理论(DFT)计算数据的系统训练和验证,确保了对Cu(111)晶面的表面能、氧吸附能以及水解离能垒的准确描述。

图1 | 铜表面氧化的原位TEM观察与ReaxFF模拟体系示意图(引自原文献Fig. 1)
2.2 模型构建
构建Cu(111)表面平板模型,包含12层Cu原子以充分捕捉表面和次表面效应。模型在x–y平面采用周期性边界条件,z方向设置真空层(约30 Å)以消除周期性镜像的虚假相互作用。
为对比水的促进效应,构建两种模拟条件:
• 纯O₂氧化条件:在Cu平板表面上引入O₂分子,模拟干燥条件下的氧化过程
• O₂ + H₂O混合条件:引入O₂和H₂O分子,模拟含水环境下的氧化过程,H₂O分子提供OH中间体
2.3 分子动力学模拟参数
所有ReaxFF分子动力学模拟使用LAMMPS程序包执行。具体参数设置如下:
• 系综:NVT(正则系综),使用Nosé-Hoover恒温器控制温度
• 温度范围:300–800 K,模拟不同热激活条件下的氧化动力学
• 时间步长:0.25 fs,以准确捕捉轻元素H原子参与的化学反应
• 总模拟时间:2–5 ns,足以观察Cu表面氧化的完整动力学过程
• Cu底层原子固定,模拟半无限大的块体约束条件
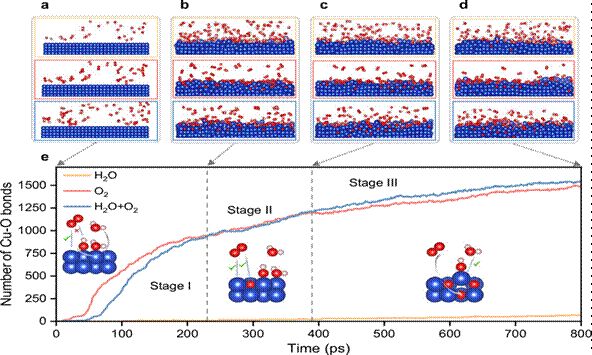
图2 | 不同氧化条件下Cu表面的原位TEM实验结果(引自原文献Fig. 2)
2.4 数据分析方法
对模拟输出的原子轨迹进行多维度分析,以量化Cu表面的氧化动力学行为:
• 原子配位数分析:统计Cu–O配位数随时间的演化,量化氧化程度
• 径向分布函数(RDF):表征Cu–O和Cu–Cu键合结构在氧化过程中的变化
• 电荷密度分析:ReaxFF通过EEM(电负性均衡方法)动态计算原子电荷,追踪界面电子结构变化
• 均方位移(MSD):计算Cu原子的迁移率,评估氧化引起的原子流动性变化
• 密度分布曲线:沿z方向统计原子密度,测定氧化层的厚度和渗透深度
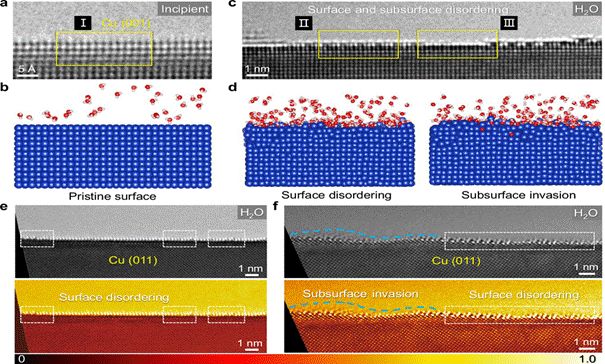
图3 | ReaxFF分子动力学模拟中Cu表面氧化过程的原子尺度快照(引自原文献Fig. 3)
三、结果解读
3.1 OH优先吸附于无序Cu/CuOₓ界面
ReaxFF分子动力学模拟显示,H₂O分子在Cu表面的解离并非随机发生。OH优先吸附于结构无序的Cu/CuOₓ界面区域,而非平整的Cu晶体表面。这种优先吸附行为源于界面无序区域Cu原子的配位不饱和状态,为OH提供了更强的结合位点。
相比之下,O₂分子在Cu表面的吸附则主要发生在结构有序的晶格位点上,无法引发如OH吸附所导致的界面重排效应。
3.2 界面结构动态无序化
模拟结果进一步揭示了Cu–OH强相互作用的独特效应:最外层Cu原子层由于与OH的强键合而产生显著的动态无序,表现为原子位置的均方根偏差(RMSD)显著升高。同时,次表面层Cu原子获得了额外的电子密度富集,形成了一种“动态无序–电子富集”的耦合效应。
这一耦合效应从根本上降低了氧原子进入Cu晶格的能量阻力,为深入氧化创造了有利的动力学条件。
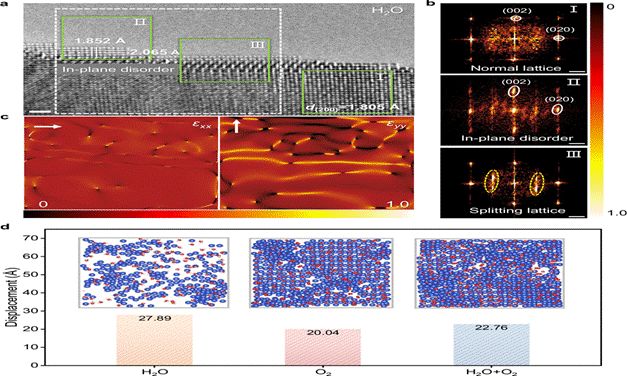
图4 | Cu/CuOₓ界面的结构分析:OH吸附引起的原子配位与径向分布变化(引自原文献Fig. 4)
3.3 水促进氧化 vs 纯氧氧化:定量对比
模拟提供了两种氧化路径的定量对比:
• 氧渗透深度:水辅助氧化条件下,氧原子可达Cu次表面3–5层;纯O₂条件下,氧主要集中在最顶层1–2层
• 氧化速率:初始0.5 ns内,水辅助条件下的O原子嵌入速率约为纯O₂条件的1.8–2.5倍
• Cu原子迁移率:MSD分析表明,水辅助条件下Cu原子的扩散系数提高了约一个数量级
纯O₂氧化形成的Cu–O有序界面实际上起到“钝化层”的作用,阻碍了后续氧的持续渗入,使进一步氧化在动力学上不利。而OH介导的界面无序则打破了这一钝化屏障。
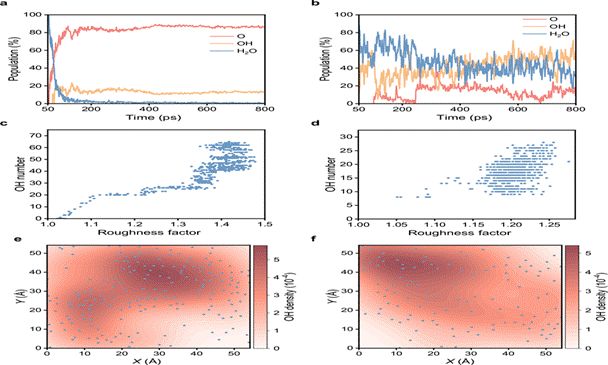
图5 | 界面电子密度分布与Cu原子迁移率的定量对比分析(引自原文献Fig. 5)
3.4 氧化机理总结
综合模拟与实验结果,论文提出了以下水促进铜表面氧化的完整机理:
① H₂O分子在Cu/CuOₓ界面上解离生成OH和H,OH优先吸附于界面无序区域的Cu位点。② Cu–OH强键合导致最外层Cu原子动态无序化,同时向次表面层转移电子密度。③ 结构无序与电子富集的协同效应降低了O的吸附和嵌入势垒。④ O持续渗入晶格深层,加速氧化并促进Cu原子迁移。⑤ 纯O₂路径因缺乏OH中间体而形成有序钝化层,氧化受到抑制。
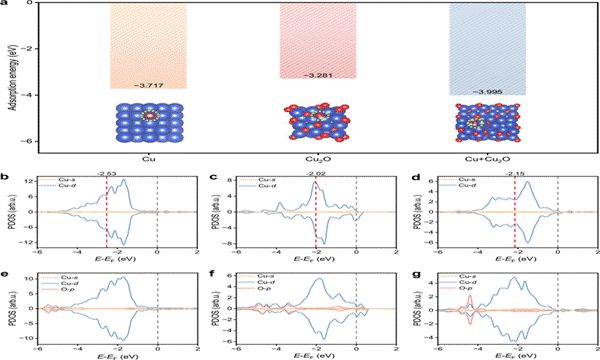
图6 | H₂O促进Cu表面氧化的OH介导界面动力学机理示意图(引自原文献Fig. 6)
四、结论与启示
该研究工作以ReaxFF反应力场模拟为核心计算手段,配合原位TEM实验验证,精确揭示了H₂O促进金属氧化的分子级机制。其科学价值在于:
• 首次在原子尺度阐明了OH中间体在金属氧化水促进效应中的具体作用路径
• 发现“结构无序–电子富集”的协同界面效应,为理解多相催化中的水效应提供了新视角
• 为设计耐腐蚀合金和优化氧化过程的反应条件提供了机理指导
该工作的研究范式——实验表征揭示宏观现象 + ReaxFF模拟解析微观机制——代表了当前材料科学研究的先进方法论。
五、方法优缺点分析
ReaxFF力场优势:
• 能够动态处理化学键的断裂与形成,无需预定义反应路径
• 基于EEM方法实时更新原子电荷,捕捉界面电子效应
• 计算效率远高于第一性原理MD(AIMD),可模拟更大体系和更长时程
局限性:
• ReaxFF力场参数的准确性依赖训练数据集的质量和覆盖范围
• 键级方法可能在某些极端条件下偏离DFT参考结果
• 适用于近平衡态反应,极端非平衡条件下的适用性需要额外验证
六、模拟计算仿真服务
我们团队可承接以下方向的模拟计算仿真项目,助力您的科研工作:
• 量子化学计算(DFT、波函数分析、反应路径搜索、激发态计算)
• 第一性原理分子动力学(AIMD、CPMD、Born-Oppenheimer MD)
• 反应力场分子动力学(ReaxFF、COMB、AIREBO,基于LAMMPS)
• 经典分子动力学(膜蛋白、材料力学、纳米摩擦、自组装)
• 有限元模拟(COMSOL多物理场耦合、Ansys结构力学、热分析)
• 相场计算(凝固、相变、晶粒生长、铁电畴演化)
• 机器学习势函数开发(DeepMD、NequIP、MACE等)
欢迎添加微信/后台留言咨询,我们提供从方案设计到数据后处理的一站式模拟计算服务!
参考文献
[1] Li S, Cao Z, Chang H, et al. Copper-hydroxyl interactions drive water-promoted copper surface oxidation and mobility. Nature Communications, 2026. DOI: 10.1038/s41467-026-70947-6
[2] van Duin ACT, et al. ReaxFF: A Reactive Force Field for Hydrocarbons. J Phys Chem A, 2001. DOI: 10.1021/jp004368u
