



快速充电电池用自适应电解液
高能电池的快速充电对交通电气化至关重要,但因电池过电位的急剧上升容易超出电解质固定的电化学稳定窗口,这一过程仍面临挑战。我们设计了一种自适应电解质,其电化学稳定窗口可实时动态扩展——随着充电电流增强,该窗口会实时扩大,从而有效抑制过电位的上升。这种自适应电解质采用盐类与互补性抗氧化/抗还原溶剂的单相溶液体系,其组成符合浊点平衡,但可通过溶剂分离实现充电过程中的溶剂成分动态重组。抗氧化溶剂在正极富集,抗还原溶剂在负极聚集,从而在充电过程中实时拓宽电解质稳定窗口。概念验证实验表明,该设计在水性锌金属电池和非水性锂金属电池中均展现出优异性能,不仅实现了负极的高库仑效率,还显著提升了正极的氧化稳定性。

文章核心创新点:
核心概念革新:引入“自适应”动态电化学稳定窗口
针对快充过程中过电位易突破传统固定电解质稳定窗口的局限,本文突破了电解质窗口恒定不变的传统设计理念,首次提出了“可实时动态调节”的电化学稳定窗口新概念。电解质如同具备智能响应能力的“防护盾”,能随充电电流的增强而自动拓宽其稳定范围,实现从静态保护到动态适应的转变。
材料体系与机制突破:依托“浊点平衡”实现单相溶液与溶剂原位分离
为达成上述动态效果,本文创新性地设计了一种由特定盐类与具备互补抗氧化/抗还原特性的溶剂组成的单相溶液。
其核心作用机制在于体系遵循“浊点平衡”原理:在充电过程中产生的电场、浓度梯度或热效应的驱动下,原本均匀的溶液会发生原位溶剂分离与动态重组,这一过程并非简单的物理混合或添加界面层。
功能实现创新:电极界面溶剂选择性自发聚集
借助上述机制,充电过程中实现了功能的精准调控:抗氧化溶剂自发在正极界面聚集,以抵御高压氧化;抗还原溶剂则自发在负极界面富集,以保护活性负极。这种“自驱动、自组织”的界面成分重构,是实时拓宽稳定窗口的直接驱动力。
性能与普适性提升:同步增强两极稳定性并验证跨体系适用性
该策略同步且显著地提升了电池两极的稳定性:既提高了负极(如锂、锌)的库仑效率,又增强了正极材料在高电位下的抗氧化性能。
概念验证覆盖了水性(锌金属电池)和非水性(锂金属电池)两大典型体系,证实了这一设计原理的跨体系普适性,为快充技术的共性难题提供了全新的通用解决方案。
总结:本文的根本创新在于,将电解质的电化学稳定窗口从传统的“固定不变的材料特性”,重新定义为能够响应外部电刺激而动态调整的“系统功能”。通过精心设计的“浊点平衡”单相溶液体系,实现了溶剂在电极界面的智能定向富集,从而为高能电池的快充挑战提供了一种前所未有的自适应应对策略。
研究背景
电解质的电化学稳定窗口是决定电池性能的关键,定义了电池无不良副反应时能稳定工作的电压区间。更宽的窗口可确保电池在更宽循环电压范围稳定运行,实现更高容量输出,拓宽它是提升电池能量密度的核心途径,从稀水溶液电解质到有机电解质锂离子电池的技术演进已印证这一点。
但进一步拓宽水系与有机电解质的窗口极具挑战。当前多数电解质稳定窗口窄,难适配高压电池或满足快充需求,而这两项是推动交通电动化的关键。为最大化能量密度,电池正、负极工作电位常处电解质稳定窗口临界边缘,只能低倍率充电,高倍率充电时过电位易超出稳定极限,引发副反应,损害电池性能。
现有降低过电位策略多集中在优化电极结构,虽能实现快充,但会牺牲能量密度。醚类电解质适配低电位负极,无法兼容高电位正极;碳酸酯类电解质正极抗氧化性能优异,却与金属锂、硅基负极不兼容。为解决矛盾,提出双相电解质概念,构建策略主要有采用固态离子传导膜物理隔离不同电解质,或将两种互不相溶溶剂混合。
然而,固态离子传导膜有脆性大、易变质、离子电导率低、成本高等缺陷,影响电池性能与经济性、安全性;互不相溶溶剂制备双相电解质也面临挑战,离子存在可能导致溶剂部分混合,充放电时离子在两相界面迁移会加剧混合。因此,研发热力学稳定的相分离电解质仍是难题。
快充另一挑战在于:电解质电化学稳定窗口固定,电池过电位随充电电流增大急剧升高,导致高电流下可用容量衰减。要快充就得采用低电压电池体系牺牲能量密度,高压电池体系无法快充。若能开发出自适应电解质,使其窗口可随充电过程实时拓宽,将为高能量密度电池电解质研发带来突破。
下一代自适应电解质需满足两项要求:一是有宽的热力学电化学稳定窗口,突破单相电解质性能局限;二是窗口可随充电电流增大拓宽。目前尚无电解质能满足这两项要求。
本研究开发了一种自适应电解质,其窗口可在充电中动态拓宽。该电解质为锂盐、耐还原溶剂与抗氧化溶剂组成的三元混合体系,组分比例精准调控使其处于浊点状态,静置时为均相体系,充电时盐析效应促使相分离,耐还原溶剂在负极侧富集,抗氧化溶剂在正极侧富集,缓解极化,拓宽稳定窗口。
该设计在水系锌金属电池与非水系锂金属电池中得到验证。本研究将溶剂相行为调控引入电解质设计体系,为突破传统电解质稳定性局限提供新范式。
研究流程
实验方法
电极与电解质制备
乙腈(ACN)等众多试剂购自阿拉丁生化科技有限公司或东京化成工业株式会社,均未进一步纯化直接使用。硒酸锌(Zn(SeO₄)₂)由氧化锌溶于硒酸溶液制备。
水系锌金属电池用自适应电解质:向乙腈/水(体积比3:7)混合溶剂中逐步加七水合硫酸锌至浊点状态;非自适应电解质组分质量分数为乙腈0.18、水0.74、硫酸锌0.08。锂金属电池用自适应电解质:向氟代碳酸乙烯酯/苯甲醚(体积比48:37)混合溶剂中逐步加双氟磺酰亚胺锂至浊点状态,其浓度约2mol/L,均在氩气手套箱(水、氧含量均低于0.1ppm)中制备。
电极制备:锌金属箔购自阿拉丁,冲压成直径16.0mm圆片用于电池组装;直径16mm锂片与厚度20μm锂箔购自美国材料技术公司(MTI);铝箔基底磷酸铁锂(LFP)电极片(面容量4.0mAh/cm²)由法国帅福得集团提供,冲压成圆片后80℃真空烘箱过夜干燥备用。五氧化二钒电极:将70%五氧化二钒、20%科琴黑与10%聚四氟乙烯(PTFE)粘结剂分散于N-甲基吡咯烷酮制成浆料,涂覆于钛箔集流体表面,80℃真空干燥12h,控制电极活性物质负载量为2.1mg/cm²。
相图绘制
采用“盐-混合溶剂”逐步混合法绘制“水-乙腈-硫酸锌”与“氟代碳酸乙烯酯-苯甲醚-双氟磺酰亚胺锂”三元相图,参照文献[31,32]方法:向盐(七水合硫酸锌或双氟磺酰亚胺锂)中逐步加不同比例混合溶剂(水+乙腈或氟代碳酸乙烯酯+苯甲醚),观察体系相态变化确定相图边界。
电化学测试
采用CR2025型扣式电池构建磷酸铁锂||锂、锂||锂、锂||铜三种测试体系。锂||铜电池用于库仑效率测试:先以0.5mA/cm²电流密度在铜电极表面沉积5mAh/cm²金属锂,随后脱锂至1V完成首次沉积/剥离循环;再沉积5mAh/cm²金属锂作锂储备层;之后以0.5mA/cm²电流密度进行10次1mAh/cm²锂沉积-剥离循环;最后将沉积锂全部脱锂至1V。全电池测试条件详见图表说明及相关讨论。为观察锂金属沉积形貌,组装锂||铜电池,以0.5mA/cm²电流密度在铜电极表面沉积5.0mAh/cm²金属锂,沉积结束后拆解电池,用乙醚冲洗电极表面备用。
表征测试
电极形貌:用日立SU-70场发射扫描电子显微镜(FE-SEM)观察。
电解质组分定量分析:用核磁共振氢谱(¹H NMR)分析充电过程中电极附近电解质组分变化。在不同电流密度下收集正负极侧电解质,加入含50mmol/L二甲基亚砜(内标)的重水中,用布鲁克AV III 600MHz核磁共振波谱仪室温采集¹H NMR谱图,通过计算特征峰面积确定乙腈含量比例。
红外光谱:用布鲁克红外光谱仪在600-4000cm⁻¹波数范围采集光谱,每张谱图累计扫描5次。
拉曼光谱:用法国HORIBA Jobin Yvon Labram Aramis拉曼光谱仪,以532nm半导体泵浦固态激光器为激发光源。
X射线衍射:用理学MiniFlex 600 X射线衍射仪,配备HyPix-400 MF二维平板探测器采集样品图谱。
计算模拟方法
密度泛函理论计算:用Gaussian 16W软件包,采用B3LYP杂化泛函与6-311++G(d,p)基组,计算不同溶剂及阴离子静电势与偶极矩。
分子动力学模拟:用大规模原子/分子并行模拟器(LAMMPS)软件,结合全原子优化液体模拟力场(OPLS-AA)开展。溶剂力场参数由LigParGen网络服务器生成,原子约束静电势(RESP)电荷通过Multiwfn程序基于静电势计算获得,硫酸锌与双氟磺酰亚胺锂分子动力学参数参照文献[42,43]设定。
模拟流程
用Packmol程序构建电解质模型初始原子坐标;用VESTA与VMD软件可视化最终结构。模拟时间步长1fs,用诺西-胡佛恒温器(时间常数0.1ps)控制体系温度293K。先在正则-等温等压系综(NPT)下对体系进行500ps平衡模拟,用帕里内洛-拉赫曼压力控制器(衰减常数1ps)维持体系压力1atm。引入退火过程:500ps内升温至363K,恒温500ps后,再用500ps降至293K。随后在正则-等温等容系综(NVT)下进行7ns模拟计算,截取最后5ns数据用于径向分布函数与均方位移分析。
结果解析
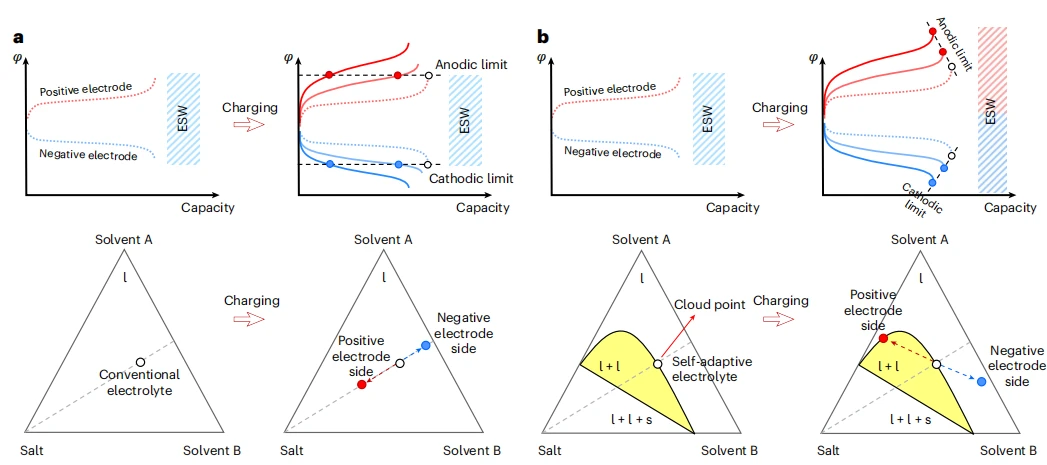
传统电解质与自适应电解质的工作原理
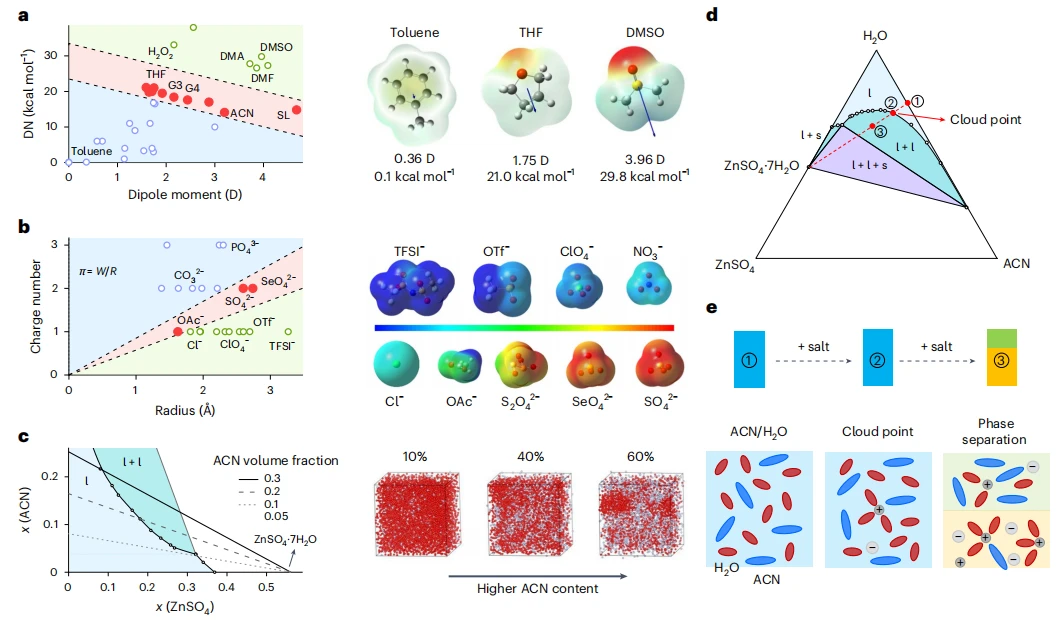
自适应电解质的设计原理
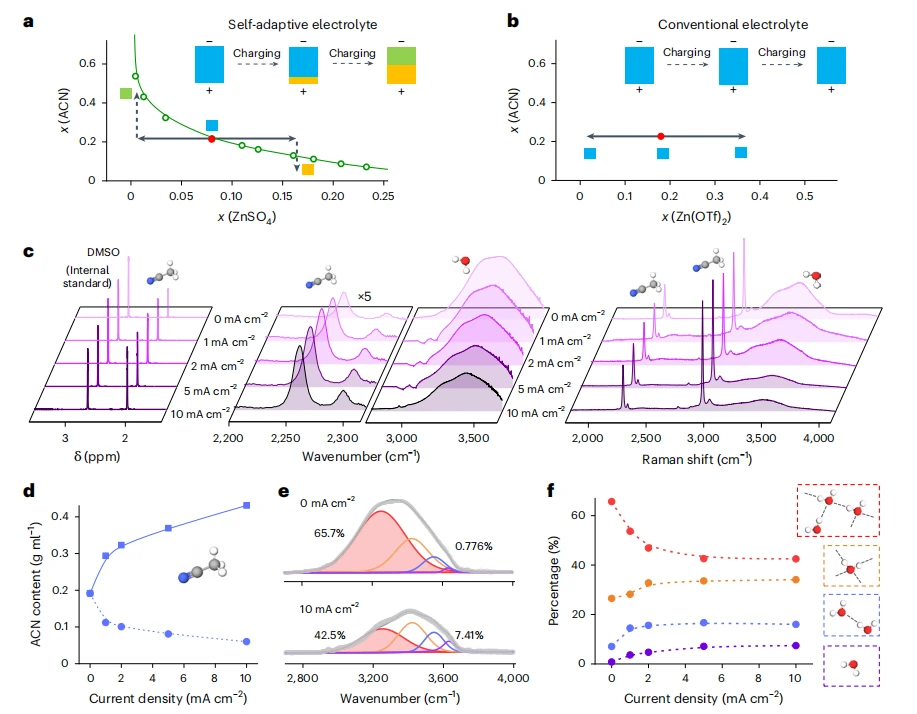
自适应电解质的电荷驱动相分离
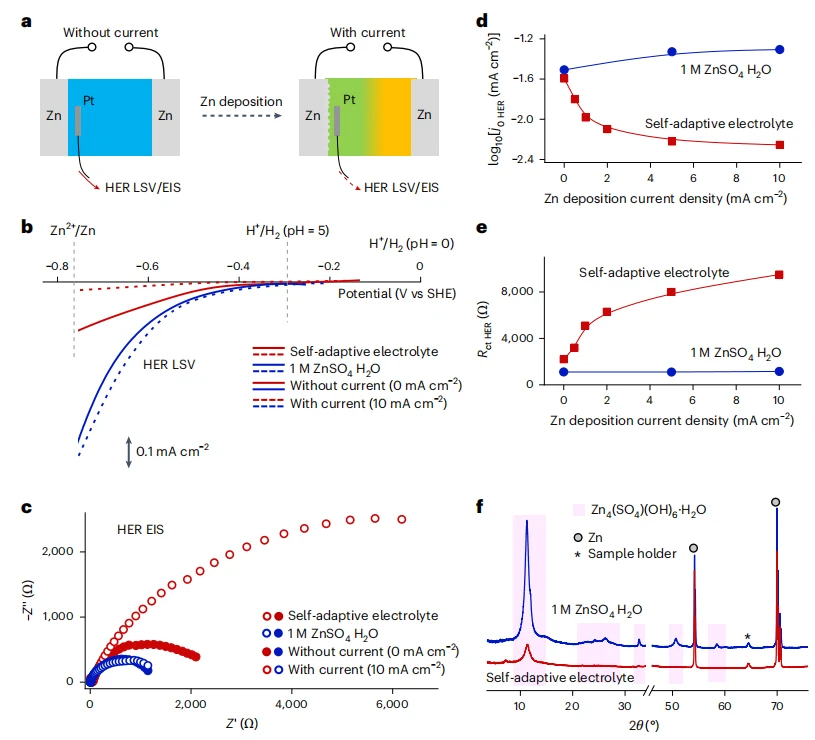
电荷驱动的自适应电解质HER抑制
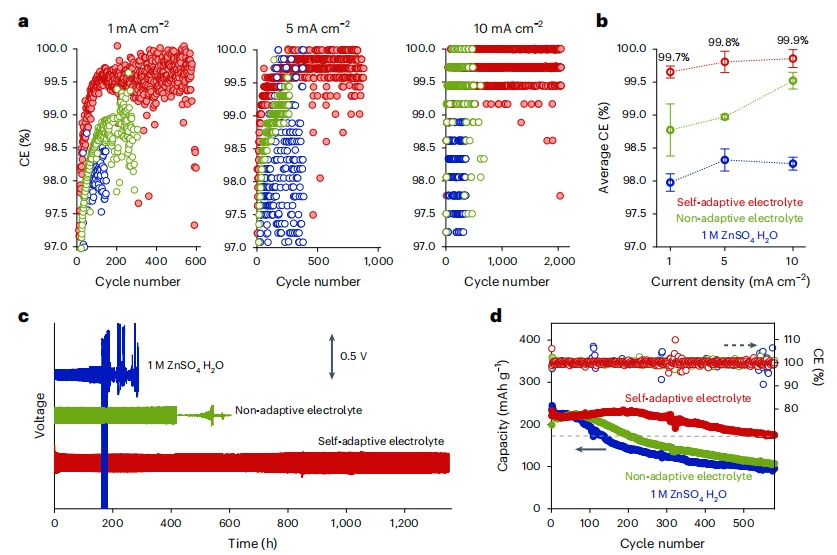
自适应电解质的电化学性能
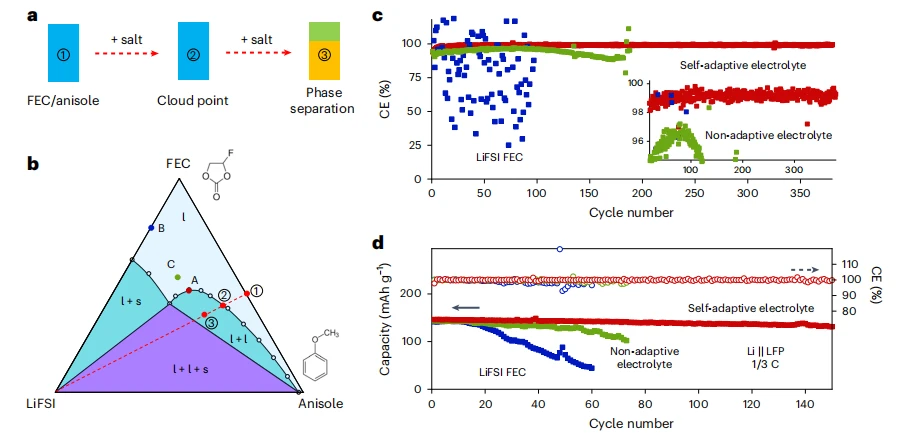
锂金属电池自适应电解质工程的普适性
研究结论
现有电池电解液具有固定的电化学稳定性窗口,这限制了高能量电池在大电流下的快速充电性能,因为过电位会迅速升高。为解决这一矛盾,我们开发了一种自适应电解液,其电化学稳定性窗口在充电过程中动态扩展。该技术采用具有溶剂排出效应的三元体系:电解液在静止时保持浊点均质状态,但充电引发的盐浓度梯度会触发相分离。这一过程使负极富集抗还原溶剂,正极富集抗氧化溶剂,从而以电流依赖的方式有效拓宽电解液的稳定性窗口。在水性锌金属电池和有机锂金属电池中均得到验证,该策略实现了快速充电而不牺牲能量密度。通过将相化学整合到电解液设计中,我们的方法超越了传统分子层面的策略,为下一代电解液建立了广泛适用的框架。
技术来源:https://doi.org/10.1038/s41560-025-01801-0
