
《Metal–organic frameworks containing N-heterocyclic carbenes and
their precursors》doi:10.1016/j.ccr.2015.06.012
原文是讲含NHC的MOF,不过里面对NHC的介绍倒也不错,翻译出来分享一下。
3.Carbenes and NHCs
卡宾碳可以是亲电的,也可以是亲核的,因此金属-卡宾物种在合成化学中有重要影响。1964年,Fischer和他的学生将卡宾引入到无机化学和有机金属化学中。他们发现的卡宾配合物具有亲电特征的卡宾碳,但是之后Schrock分离得到的卡宾配合物却含有亲核特征的卡宾碳。后来,Öfele、Wanzlick和Schönherr独立地报道了第一例过渡金属络合物,他们独立地通过使用碱性的金属前体对咪唑盐去质子化得到imidazolin-2-ylidene络合物。
在很长一段时间内,卡宾被认为是反应活性过强且短寿命的分子,不可能在不二聚的情况下分离得到。在1991年Arduengo等人分离得到稳定的、结晶的NHC之前,人们对使用卡宾作为配体的兴趣有限。NHC是一个单线态卡宾,卡宾碳被结合在含氮的杂环中。第一例稳定NHC含有基于imidazolin-2-ylidene环的骨架,并在氮原子上具有金刚烷取代基。它在没有氧气和水分的情况下具有热力学稳定性和动力学稳定性。人们最初认为该结构中体积很大的金刚烷取代基起到了保护作用,阻止了它二聚,但后来又有人分离得到了氮原子上取代基体积远小于金刚烷基的NHCs,证明了空间位阻并不是解释首例卡宾稳定性的唯一因素。

在该例分离之后,对NHCs的合成、相应金属络合物的制备有了大量的研究,并被应用于金属基催化剂领域。此外,Enders等人还对NHCs的有机催化应用进行了综述。最常见的氮杂环卡宾都含有咪唑环。它们富电子的杂环为稳定卡宾中心提供了合适结构。一般来说,NHC配体是通过用强碱,如叔丁醇钾,对N,N’-二取代咪唑盐(N,N’-disubstituted imidazolium salt)(或其他咪唑盐azolium)脱质得到的。因此,含氮配体,例如咪唑、三唑、苯并咪唑(imidazolium, triazolium,benzimidazolium)是生成NHC配体的前体。除了对氮盐(azolium)脱质子外,还有一些不太常用的生成游离卡宾的方法。
NHCs具有很强的σ-给体能力以及较差的(poor)π-受体能力,这导致了它们的化学稳定性及配位的多样性,从而它们可以和多种金属(从碱土金属到过渡金属)配位。此外,将手性元素引入NHCs非常容易并且前体的制备简便,使得手性NHCs在金属基不对称催化中成为很有前景的手性配体。值得注意的是,NHC衍生的第二代催化剂相较于第一代催化剂,不仅对烯烃复分解有更好的催化活性,并且可以通过变化取代基对催化活性进行微调。这可以从Fig. 4的2和4所示的Grubbs第二代催化剂中观察到。

3.1. Stability and reactivity
不同于三线态卡宾具有类似于自由基的反应性,单线态卡宾的σ型孤对电子和空p轨道分别使它表现出亲核反应性和亲电反应性。单线态卡宾可以分为五种:+M/+M, -M/-M, -M/+M, +M/-以及最近分离得到的芳香环丙烯(aromatic cyclo-propylidenes)。这种分类方式是基于中介效应(mesomeric effects, ±M)以及α-基团带来的稳定化作用。NHCs属于+M/-基团,它同时被p轨道取代基向亚甲基空的p轨道的给予以及卡宾填充的p轨道对取代基空的p轨道的弱给予所稳定,这组成了一个推拉效应(NHCs belong to the +M/- group, which is stabilized simultaneously by a strong donation from a p-orbital substituent to the methylene empty p-orbital and by a weaker donation from the carbene’s filled p-orbital to the substituent’s empty p-orbital, thus constituting a push–pull effect)。π-给予对游离NHC稳定性的贡献较少,因为大量的从卡宾碳到电负性更强的邻位氮原子的σ电荷转移对稳定性做出了主要的贡献。从中介效应来看(不确定怎么翻译,Mesomerically),作为单线态卡宾的NHCs倾向于形成弯曲的几何结构。
空间稳定的imidazolidenes和imidazolylidenes在一定程度上是热力学稳定的,它们不与三线态氧气反应,甚至形成酮也是大量放热的(Sterically stabilized imidazolidenes and imidazolylidenes are thermodynamically stable to the extent that they do not react with triplet dioxygen, even when the formation of the ketone appears to be very exothermic)。尽管Arduengo课题组后来通过将大位阻的金刚烷取代基换成更小的甲基,得到了立体位阻更小的稳定的imidazol-2-ylidene,不过空间位阻因素仍对NHCs的稳定性和反应性有贡献。Denk等调查了1,3-di-tert-butylimidazol-2-ylidene和1,3-di-tertbutylimidazolin-2-ylidene对H2, O2, H2O及CO的反应性。他们报道了NHCs并不与O2或CO反应,但是对空气敏感,因为它们会被H2O进攻,从而开环生成甲酰胺(formamides)。在C4和C5上有不饱和骨架的NHCs在水中比含饱和骨架的NHCs更稳定。这些卡宾通过被OH-亲核进攻或插入H2O中水解,历经环状α-二胺基甲醇(cyclic -diamino methanol)中间体开环得到甲酰胺。此外,及时长时间加热,它们也不与氧化剂(如CuO和HgO)反应。在铂作为催化剂存在的条件下,NHCs与氢气反应生成1,1-加氢产物(1,1-hydrogenation)缩醛胺(aminals)。
NHCs表现出一种矛盾的p-键合特征,既起π-酸作用又起π-碱作用。这种双重行为在二聚中表现得很明显,二聚反应被认为是最简单的卡宾反应。对于低d电子数如d0体系,π-给于及π-反馈都很重要。但在含高d电子数的体系中p-相互作用主要为反馈作用。因此在高d电子数体系中NHC配体最好被看作π-酸。
值得注意的是,C4和C5上的取代基以及N-C-N键是决定NHCs碱性的主要因素。它们的碱性不是很依赖电子离域。因此,非环状双(二异丙基氨基)卡宾(acyclic bis(diisopropylamino) carbene)配体以及C-C饱和的dimesitylimidazolidin-2-ylidene相比于C-C不饱和的N,N-di-(R)-imidazolin-2-ylidenes能在金属原子上引入更高的电子密度。NHCs与酸反应形成亚胺盐(iminium salts),它们是最强的中性有机碱之一。
NHCs的二聚包括了一个卡宾中心的σ孤对电子对另一个卡宾中心的空p价轨道的进攻。基本上,二聚的热力学趋势与空间和电子性质相联系。适当增加氮原子上R取代基的空间位阻有利于形成单体NHC,但位阻过大的NHC配体对催化一般是有不利影响的。为了对NHCs的二聚在电子效应和空间效应上建立平衡,Poater等提出了一个方程,有助于校准这两个效应。π-离域是含有不饱和骨架NHCs的芳香性和一般NHCs稳定性的先决条件。令人惊讶的是,两个具有高于氮和硫电负性的氧原子提供的稳定化作用最低。这表明尽管不饱和NHCs骨架中的双键对它的稳定性有贡献,但两个氮原子向卡宾碳的pπ-pπ电子给予才对卡宾的稳定性有主要贡献。
imidazole-2-ylidene的C4和C5基团是酸性的,它们的氢原子被卤素或氘取代就是例子。一种非常空气稳定的NHC,1,3-dimesityl-4,5-dichloroimidazol-2-ylidene,就是通过将1,3-dimesitylimidazol-2-ylidene的C4和C5氢进行氯代得到的。氯代NHC的高稳定性可归因于氯孤对电子的π-给电子作用及氯高电负性带来的σ-吸电子作用。这降低并稳定了卡宾中心σ孤对电子的碱性。此外,由于骨架的酸性,NHC的C4和C5可是被锇取代。
4. NHC-metal bonds
对NHC与金属成键的理解对研究者来说是一个有启迪性的经历。尽管NHC基催化剂(NHC-based catalysts)具有卡宾官能团,但它们在许多催化条件(如温度、压力)下非常稳定。这种能力引起了学术和工业上的研究,于是,大量有效的、具有不同结构的含NHC的催化剂被合成出来,其中一些比它们的膦基类似物催化能力更好。一般来说,NHC-金属络合物可以通过直接金属化或转金属化合成。后者一般包括用Ag2O处理咪唑盐形成Ag-NHC络合物,随后用所需的金属盐进行转金属化,例如,用Ag-NHC络合物转金属化生成双(NHC)-Pd络合物。NHCs的一般结构和其配合物的一些例子如Fig. 5所示。NHCs的扁平杂环结构呈扇形,由取代基R1、R2和金属原子M组成,它们可能可以分别沿着R1-N、R2-N和C-M键旋转。NHCs的强σ-给体和弱的π-受体能力使它的金属络合物在非常多的反应中都是很好的催化剂,包括加氢酰化、Suzuki-Miyaura反应、氢化和钌基烯烃复分解反应。Herrmann等报道了NHC作为配体在过渡金属催化中的首例应用,Díez-González等对NHC-后过渡金属络合物的应用进行了详细地综述和分类。除了催化,Mercs和Albrecht总结了NHC络合物的其他应用。
4.1. NHCs as σ- and π-donors and as π-acceptors
在NHC-金属键合中,电子效应和空间位阻效应同样重要。含NHC前体的配体在实际应用中的重要需求促使了大量从实验上到理论上有关理解NHC-金属键的工作进行。NHC-金属键的三个轨道贡献如Fig. 6所示。
NHC配体在通过Fig. 6a所示的σ→d给予与金属离子配位时,被认为是简单的纯σ-给体。NHCs可与主族元素、稀土金属(如铝、镁和铊)形成稳定加和物,因为NHCs具有纯σ-给体特征并且这些金属中心无法形成π-反馈(注:此处原文写的就是rare-earth metals)。铍离子是Lewis酸性金属离子,可以与卡宾1,3-dimethylimidazolin-2-ylidene形成稳定加合物,进一步佐证了NHC的纯σ→d给予,因为铍的案例中不可能有反馈作用。这种络合物的稳定性被卡宾碳原子上取代基(即NHCs的氮原子)对纯受体金属铍的给予作用所增强。然而,在研究Mg-C(NHC), Ca-C(NHC), Ba-C(NHC)络合物中的M-C(NHC)距离时,发现越轻的碱土金属具有越强的σ-相互作用。此外,Mungur等指出金属-卡宾键的强度在本质上主要是静电性质的。他们合成了首个s区金属(锂和镁)和铀酰胺(uranium amido)NHC络合物,发现它们的金属-卡宾键与预期的平面三角形杂化相比有明显的变形,但键的强度并没有降低。Pd-NHC络合物通过它们相应的咪唑卤化物盐在K2CO3作为碱的存在下与PdCl2和吡啶反应合成(Pd–NHC complexes were synthesized from their respective imidazolium halide salts by the reaction of PdCl2 with pyridine in the presence of K2CO3 as a base)。通过电荷分解分析考察络合物中NHC-金属键的性质,发现NHC配体是有效的σ-给体。
另一方面,认为NHCs只是纯σ-给体的观点已经改变了,因为这些配体的电子性质要比预期的灵活得多。卡宾上填满或空的π-轨道及NHC环都参与到了与金属的成键中。
由于NHC配体的灵活性,它们可以通过π→d给予稳定缺电子金属或通过d→π*反馈稳定富电子金属。

1998年,Boehme和Frenking报道了第一例微弱金属→配体π-反馈成键作用,d→π*给予(Fig. 6b)。Lee和Hu通过使用密度泛函理论研究了Cr(CO)5L络合物,其中L为NHCs、膦基(phosphines)、二氨基卡宾(diaminocarbenes)、Fischer和Schrock型卡宾。他们观察到NHCs的配体-金属键远强于膦基的配体-金属键,并且在NHC-金属键中,π-反馈作用与σ-给予作用相比可以忽略不计。
在结构信息的基础上,卡宾配体和金属间的强π-金属相互作用已被报道。Hu等利用最近发展的现代DFTs和相关计算机程序计算了合成的NHC-金属络合物的电子结构,他们证明了NHCs可以通过d→π*反馈的方式从11族富电子金属原子接受电子密度。对11族金属与NHC配体络合物的能量分解分析计算得到了相似的结论。实验和计算结果表明,NHC在与富电子的U(III)离子配位时可以作为π-受体配体。Jacobsen等比较了形式d电子数从d0到d10的一系列NHC络合物中σ-和π-的贡献。他们报道即使是d0络合物也存在相当数量(considerable)的金属→配体π-反馈。此外,他们指出在高d电子数的体系中反馈作用是π-相互作用的主要贡献者。使用了傅里叶变换红外光谱和循环伏安法两种技术来观察C4和C5位连有对醌结构(p-quinone moiety)的NHC络合物的结构和电子变化,包括σ-和π-效应(deconvoluting σ- versus π-effects)(见Fig. 7)。

并在咪唑亚基(imidazolylidene)上的对醌结构对π-效应非常敏感,很适合识别金属络合物的π-反馈键,而顺,顺-1,5-环辛二烯配体与两单位的CO迅速交换(The p-quinone moiety fused to the imidazolylidene is sensitive to π-effects and is ideal for identifying π-backbonding of the metal complexes, while the cis,cis-1,5-cyclooctadiene ligand rapidly exchanges with two units of CO)。通过分析,观察到了大量的π-反馈作用。
在研究了NHC-金属键的σ-给予作用和π*-反馈作用后,发现σ-给予作用远强于π*-反馈作用,计算出的σ/π*比值远大于对经典Fischer卡宾化合物的计算值。尽管π*-反馈在硅宾、锗宾和其他卡宾化合物中略强一些,但这种作用基本上可以忽略不计。值得注意的是,NHC-金属键主要是由于卡宾原子上孤对电子和金属原子上的正电荷的库伦引力,但同时也有显著的给体孤对电子的共价相互作用。电荷分解分析主要确认了卡宾配体几乎纯的σ-给体性质。用DFT对一系列结构不同的银和金的酰胺功能化(amidofunctionalized)的NHC络合物的NHC-金属键合本质进行研究,结果表明NHCs是好的σ-给体配体及其相对弱的π-受体性质。NHCs卡宾的孤对电子填充到了金属的一个空p或s轨道上(Ag的5p或5s,Au的6p或6s)。
对合成的Pt(II)-NHC络合物的电子密度分析表明,与不饱和NHC类似物(1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene和1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene)相比,金属中心对饱和NHCs(1,3-bis(2,6-diisopropyl)phenyl-4,5-dihydroimidazol-2-ylidene和1,3-bis(2,4,6-trimethyl)phenyl-4,5-dihydroimidazol-2-ylidene)反馈的电子密度更多。此外,饱和NHCs与不饱和类似物相比对Pt(II)有更多的σ-给予。因此,由于π-反馈作用和σ-给予作用的协同效应,饱和NHC体系中Pt(II)的电子密度要高于不饱和NHC体系。所以,饱和NHC与金属的结合要比相应的不饱和NHC更强。这些观察结果很大程度上解释了为什么不饱和NHC催化剂与底物的配位更容易,尽管它们的稳定性被其与金属较弱的键合所影响。相反地,饱和NHCs是结合能力更强的配体,它可以稳定催化剂但也可能降低催化剂的反应性,这可能是因为它们将金属中心变成了一个差的σ-受体和π-给体,导致其与底物的结合变弱。
NHCs不仅通过NHC-金属键合接受金属的π电子,它们也可以向金属给予π电子。Jacobsen等用DFT和适当的键分析表明对于具有低d电子数目的体系,π-给予和π-反馈对于NHC-金属键合都是重要的。为了解释IrCl(ItBu)2 (其中ItBu为1,3-bis(tertbutyl)imidazol-2-ylidene)中不存在不可知的相互作用(agnostic interactions),Cavallo等报道了NHC配体π-轨道的给予作用相比于叔丁基C-H键σ-轨道的给予作用能更好地补偿金属络合物的缺电子性。此外,Scott等通过分子轨道分析报道了14-电子络合物[Ir(ItBu)2]PF6的异常稳定性是由于NHC配体填充的π分子轨道通过显著的π→d给予向铱原子空的d轨道注入了电子密度。
4.2. Steric effect
一般地,在给定配位环境中NHC配体电子和空间性质的结合会影响卡宾孤对电子的稳定性。从结构的角度来看可得出结论:连在NHC配体氮原子上的大位阻基团和金属络合物中短的金属-碳间距都会为金属中心带来比三级膦基(tertiary phosphines)更拥挤的空间位阻。
计算了Ni(CO)3(NHC)体系中CO的键离解能(BDE),以解释两类NHC基配合物(饱和NHC配合物和不饱和NHC配合物)表现出的不同行为。这两类NHC配体的差异完全归因于空间因素。1,3-Bis(adamantyl)imidazol-2-ylidene和ItBu比很大的膦基配体和其他NHC配体还要大得多。
位阻效应不仅在决定NHC-金属键的BDE中起重要的作用,也可以完全改变络合物的稳定性。Cavallo等报道在Cp*Ru(NHC)Cl(其中Cp*为η5-C5Me5)类络合物中,NHC配体的BDE明显依赖配体芳环邻位上取代基的大小。然而,在Ni(CO)3(NHC)类络合物中,稳定性则依赖于连在NHC配体氮原子上的基团的大小。对于较小的1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene基和 1,3-bis(2,4,6-trimethyl)phenyl-4,5-dihydroimidazol-2-ylidene基配体,饱和的Ni(CO)3(NHC)是稳定的;然而对于大体积的1,3-bis(adamantyl)imidazol-2-ylidene基和ItBu基配体,18-电子的Ni(CO)3(NHC)却极不稳定,形成16-电子的Ni(CO)2(NHC)络合物。
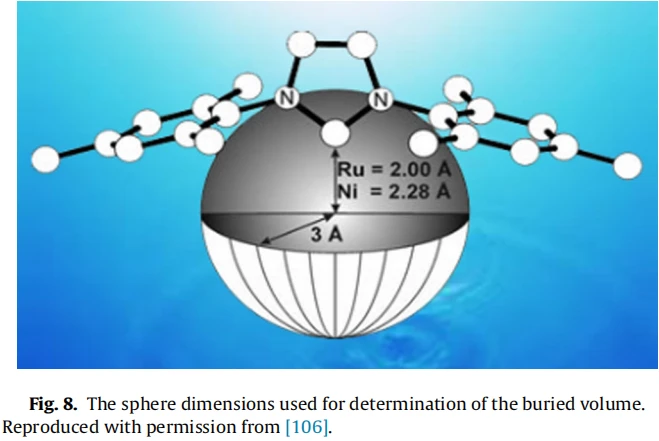
百分埋藏体积(percent buried volume,以金属为中心的球体内配体原子所占体积的百分比)的概念被用于量化NHC和膦基配体的空间位阻大小(Fig. 8)。该分子描述符可被认为是三级膦基Tolman锥角(percent buried volume)的近似物。这个球形的体积代表了金属原子配位时周围必须被不同配体所共享的空间。
基本上,配体越大,它所占据的球形空间就越多,即埋藏体积就更大。假设将金属原子钌和镍分别放在距离配位碳2.00Å和2.28Å的位置上,并用Fig. 8中的球体来确定不同配体的体积。虽然BDE本质上是由配体的空间需求控制的,但埋藏体积可以展示出不同配体的不同空间需求。对于含有不同烯基部分的(NHC)Pd(R-allyl)Cl络合物的空间和催化研究表明,含有饱和的2,6-二异丙基苯基(2,6-diisopropylphenyl (SIiPr))络合物,其进行芳基氨化反应(aryl amination)的速度远快于其不饱和IiPr类似物,这是因为饱和SIiPr配体拥有更高的空间位阻。然而,金属中心周围过大的位阻压力会导致NHC-金属键不稳定。
