

作者:我肛肛好
摘 要:通过蛋白质的氨基酸序列来预测其三、四级结构是困扰科学家几十年的难题,而Alphafold和RoseTTAFold的开源,从一定程度上为解决该难题提供了参考方案。然而目前人工智能在生物医药领域应用的案例较少且时间尚短,所以Alphafold和RoseTTAFold对蛋白结构的预测准确性值得评估检验。本文旨在评估Alphafold和RoseTTAFold预测蛋白模型的质量以及与晶体结构比较的相似度。本文选择开源后提交在PDB数据库中已具有较高同源性蛋白结构的3个蛋白和具有低同源性蛋白结构(低于30%)的3个蛋白,分别使用拉氏图评估预测模型的质量,再将预测模型与晶体结构进行比对,评估模型的准确性。结果显示,Alphafold和RoseTTAFold在预测已具有较高同源性蛋白结构的蛋白好于具有低同源性蛋白结构的蛋白,Alphafold在两种情况下的预测准确性都高于RoseTTAFold。该结果为将来生命科学领域的人工智能软件研究发展提供参考。
关键词:人工智能;蛋白质结构预测;Alphafold;RoseTTAFold;准确性评估
Study on the accuracy of protein structure prediction with Alphafold and RoseTTAFold
Abstract: Predicting the tertiary and quaternary structure of a protein by its amino acid sequence is a problem that has plagued scientists for decades. The open source of Alphafold and RoseTTAFold provides a reference solution for this problem to a certain extent. However,there are few cases of artificial intelligence application in the field of biomedicine and it just appeared soon,so the accuracy of protein structure prediction with Alphafold and RoseTTAFold is worthy of evaluating. This article aims to evaluate the quality of protein models that were predicted by Alphafold and RoseTTAFold and compare protein models with the crystal structure. Three proteins with high homologous protein structures and three proteins with low homologous protein structures (less than 30%) in the PDB database were selected to evaluate, which were deposited into the PDB database after the open source. The Ramachandran Plot was used to evaluate the quality of the predicted models, and then the predicted models were compared with the crystal structures for evaluating the accuracy of the models. The results show that the models predicted by Alphafold and RoseTTAFold with high homologous structures are better than that with low homologous protein structures. The prediction accuracy with Alphafold is higher than that with RoseTTAFold in both cases. The results provide a reference for the research and development of artificial intelligence software in the field of life sciences.
Keywords: artificial intelligence; protein structure prediction; Alphafold; RoseTTAFold; accuracy evaluation
蛋白质是生命的物质基础,每个蛋白质的氨基酸链经过扭曲、折叠、缠绕成复杂的结构[1,2]。截至目前,已有约17万蛋白质的结构经实验解析,提交至蛋白质数据库(Protein Data Bank,PDB),但这些蛋白质只占很小一部分。蛋白质结构为很多的致病机制、药物研发等提供基础。在蛋白质结构解析的几十年历史中,X射线晶体学[3]、核磁共振(NMR)[4,5]和冷冻电镜(Cryo-EM)[6]技术发挥了巨大的贡献,但这些技术通常耗时长、成本高。
由此,高效地通过蛋白质的氨基酸序列来预测其结构具有重大意义。谷歌旗下人工智能公司DeepMind在去年12月的国际蛋白质结构预测竞赛CASP上夺得头筹[7, 8],他们开发的基于神经网络的AlphaFold2击败了其他选手,预测准确性接近实验结果。并于7月,DeepMind团队在《自然》(Nature)发表相关论文[9],首次对外分享开源代码;与此同时,David Baker领导的研究组开发出一款名为RoseTTAFold的工具,发表在《科学》(Science)上,其基于一个“三轨”神经网络只需较低处理能力的计算机就能根据有限的信息快速准确地预测出目标蛋白质的结构[10,11]。
本文旨在研究AlphaFold和RoseTTAFold预测蛋白结构时的模型质量,并比较预测模型与晶体结构的相似度。选择开源后提交在PDB数据库中已具有较高同源性蛋白结构的3个蛋白和具有低同源性蛋白结构(低于30%)的3个蛋白,分别测试Alphafold和RoseTTAFold在有、无同源性结构模板情况下预测模型的准确程度[12]。首先通过拉氏图(Ramachandran plot)来评估预测蛋白模型的质量,再通过TM-Score,RMSD及Contact Map Overlap比较预测模型与相应晶体结构的相似程度,从而综合评估预测模型的准确性。
1 实验与方法
1.1 蛋白质结构的选择
本文选择开源后提交在PDB数据库中已具有较高同源性蛋白结构(>30%)的3个蛋白(PDB编号分别是7LA7,7M5I,7ONO),和具有低同源性蛋白结构(<30%)的3个蛋白(PDB编号分别是6Z5Y,7AM1,7LT2),分别测试Alphafold和RoseTTAFold预测模型的质量及与晶体结构比较的相似性。
选择开源后提交在PDB数据库中具有低同源性蛋白结构的蛋白是为了阻止Alphafold和RoseTTAFold在离线结构数据库中直接匹配到结构模板。
1.2 预测模型质量的评估
在评估预测模型的准确性前,判断模型本身的合理性是必要的,本研究使用拉氏图对模型质量进行评估。
拉氏图表示的是α碳的两面角,φ(phi)表示一个肽单位中α碳左边C-N键的旋转角度,ψ(psi)表示α碳右边C-C键的旋转角度。理论上C-N键和C-C键都可以自由的转动,由于键的转动会带动其它原子的一起转动, phi(CN-CalphaC)和psi(NCalpha-CN)较为自由,而肽平面是刚性的omega(OC-NH)通常是180°。在实际中由于分子各个基团的空间障碍和作用力的影响,拉氏图就产生了氨基酸构象允许的区域和不允许的区域[13,14]。
1.3 与晶体结构比较预测模型相似性的评估
均方根差(RMSD)表示预测模型与晶体结构中相应的原子在最佳刚体叠加后的平均距离。尽管RMSD是一个广泛的评分标准,但只有在差异很小且分布均等的情况下才有意义。[15-17]同时,本研究采用TM-score来衡量预测模型与晶体结构之间的相似性。TM-score作为全长蛋白质结构全局相似性的更准确度量,其通过一个分数来表示两个结构之间的相似性(0,1],其中 1 表示两个结构完全一致(http://zhanggroup.org/TM-align/)。[19, 20]
Contact map plot描述了蛋白质中残基的成对空间和功能关系[21]。Contact map plot是一个二进制、正方形、对称矩阵,是蛋白质 3D 结构的二维表示。本文运用了尼德曼-翁施(Needleman-Wunsch)全局对齐算法[22]实现迭代查询-模板对齐。残基对的Cα或Cβ原子之间的空间距离小于给定的距离阈值则判断为接触,本文设置为8 Å。通过Contact Map Overlap查找预测模型接触图与晶体结构接触图之间的相似性,并给出百分比分数,分数越高表示两个比较接触图之间的相似性越高。
2 结果
2.1 预测模型质量评估
拉氏图检合格的模型应该有超过90%的残基都应该出现在最佳允许区域和允许区域内。
2.1.1 已具有较高同源性结构的蛋白7LA7,7M5I和7ONO的拉氏图
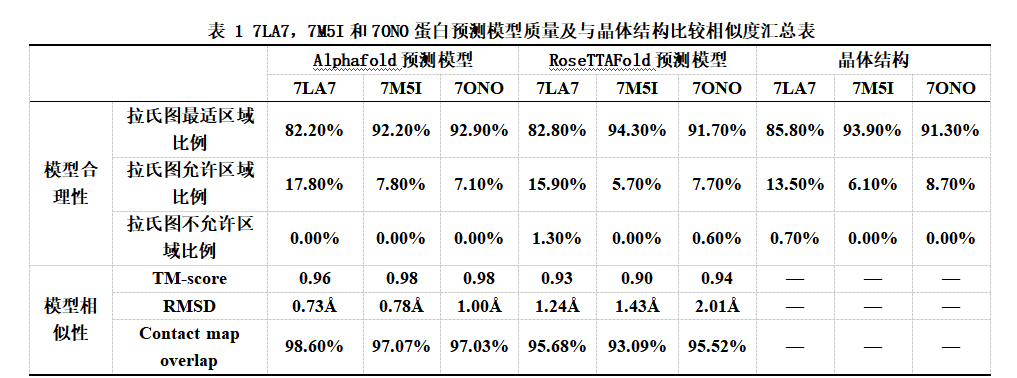
由于7LA7晶体结构位于拉氏图最佳区域的残基数比例较低,其在拉氏图中最佳区域的残基比例仅占85.80%,在允许范围区域的残基比例仅占13.50%,出现在不允许区域的残基数比例占0.70%(图1.C,表.1);Alphafold 和RoseTTAFold预测模型在拉氏图的评估下质量一般。Alphafold预测模型中出现在在拉氏图最佳区域的残基比例为82.20%,出现在允许区域的氨基酸残基比例为17.80%,出现在不允许区域的残基占0%;RoseTTAFold模型中出现在在拉氏图最佳区域的残基数比例为82.80%,出现在允许区域的氨基酸残基比例为15.90%,出现在不允许区域的氨基酸残基比例为1.30%。
在7M5I和7ONO中,Alphafold 和RoseTTAFold预测模型的质量都较好。Alphafold的两个模型在拉氏图中最佳区域的残基比例分别为92.2%和92.9%,出现在拉氏图的允许区域的残疾比例分别为7.80% 和7.1%,而出现在拉氏图的不允许区域的残疾比例皆为0(图1.D-I,表.1)。RoseTTAFold的两个模型的氨基酸出现在拉氏图中最佳区域的残基比例分别为94.3% 和 91.7%,允许区域的残基比例分别为5.7% 和 7.7%,而不允许区域的残基比例分别为0和 0.6%。在7M5I和7ONO两个蛋白的预测中,两个软件的预测模型质量都较好,RoseTTAFold模型中氨基酸残基出现在拉氏图最佳位置和不允许位置上都比Alphafold模型稍多一些。
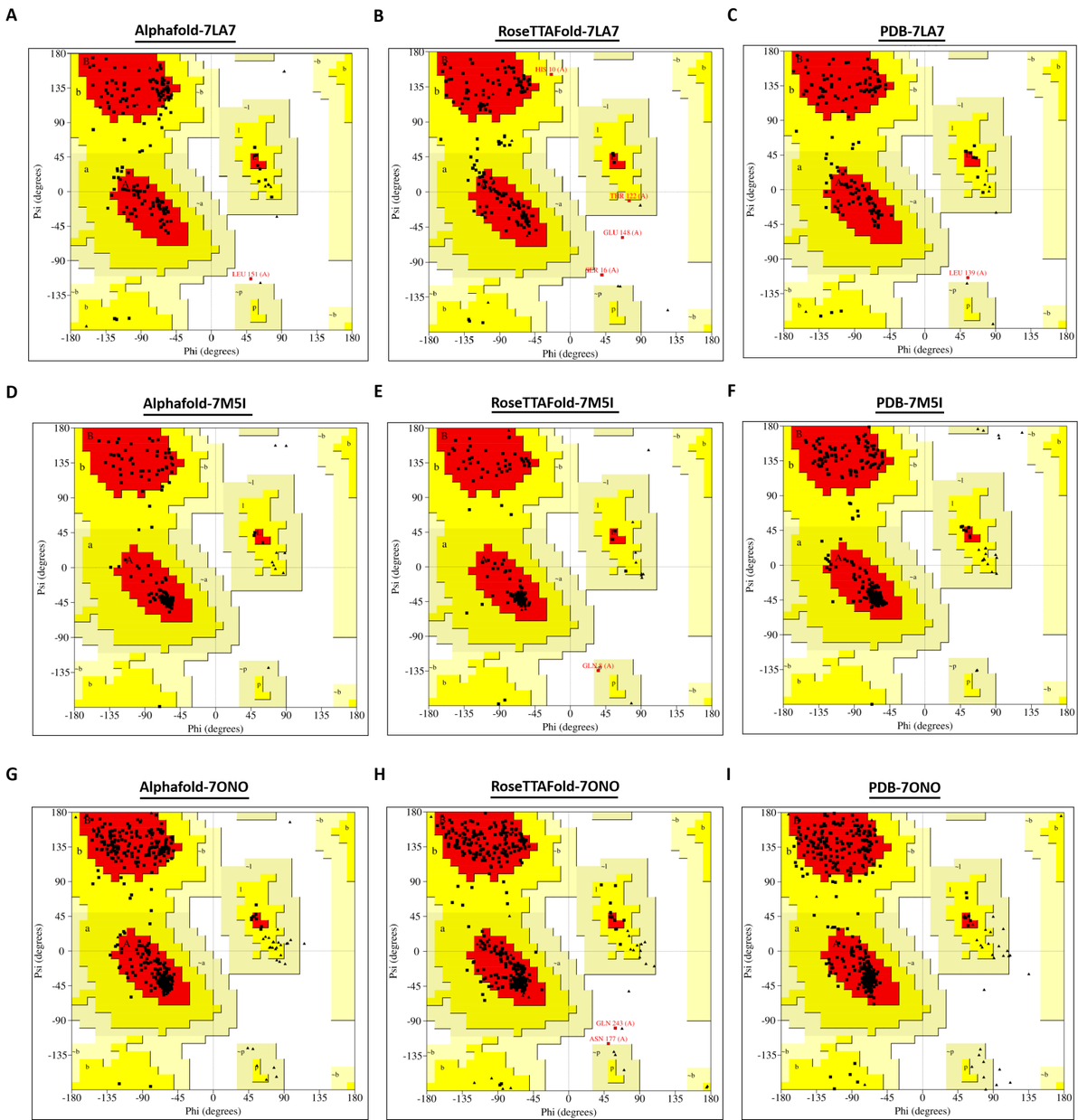
图 1. 蛋白7LA7,7M5I和7ONO的拉氏图
2.1.2 已具有低同源性蛋白结构的蛋白的预测模型与其对应晶体结构比较
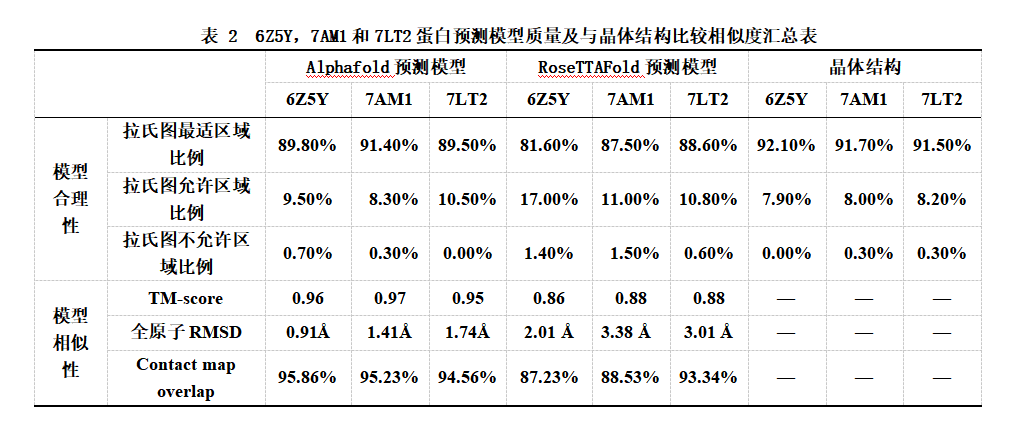
蛋白6Z5Y的Alphafold预测模型位于拉氏图最佳区域的残基数比例为89.8%,位于拉氏图允许区域的残基数比为9.50%,位于拉氏图不允许区域的残基数比例0.70%,相比之下,蛋白6Z5Y的RoseTTAFold预测模型中,位于拉氏图最佳区域的残基数比例为81.6% 低于90%,位于允许区域的残基数比例17.00%,位于不允许区域的氨基酸残基比例为1.40%,可见其模型质量上可能存在些许问题,因为在6Z5Y晶体结构中没有一个氨基酸残基位于不适合区域,同时拉氏图最佳区域的残基数比例为92%。
在7AM1的晶体结构中,从81位谷氨酸(E)~150位天冬氨酸(N)的结构是缺失的,而Alphafold和RoseTTAFold分别预测这段缺失的结构为一段自由活动的无规则结构,和几段α螺旋结构(图.6 B)。为了排除缺失段的干扰,这里做了align后的拉氏图。Alphafold预测模型位于拉氏图最佳区域的残基数比例为91.40%,出现在被允许区域的氨基酸残基比例为8.30%,在不被允许区域的比例为0.30%;RoseTTAFold预测模型位于拉氏图最佳区域的残基数比例为87.50%,出现在被允许区域的比例为11.00%,在不被允许区域的比例占1.50%。预测模型质量上,Alphafold要略好于RoseTTAFold。
在7LT2预测模型中,Alphafold预测模型位于拉氏图最佳区域的残基数比例为89.50%,出现在被允许区域的氨基酸残基比例为10.50%,出现在不被允许区域的氨基酸残基的比例占0%;RoseTTAFold预测模型位于拉氏图最佳区域的残基数比例为88.60%,出现在被允许区域的氨基酸残基比例为10.80%,不被允许的比例占0.60%(图.2)。
总的来说,在蛋白结构同源性较低的情况下,Alphafold和RoseTTAFold预测模型的质量有所下降,其中RoseTTAFold的模型质量下降较为明显。
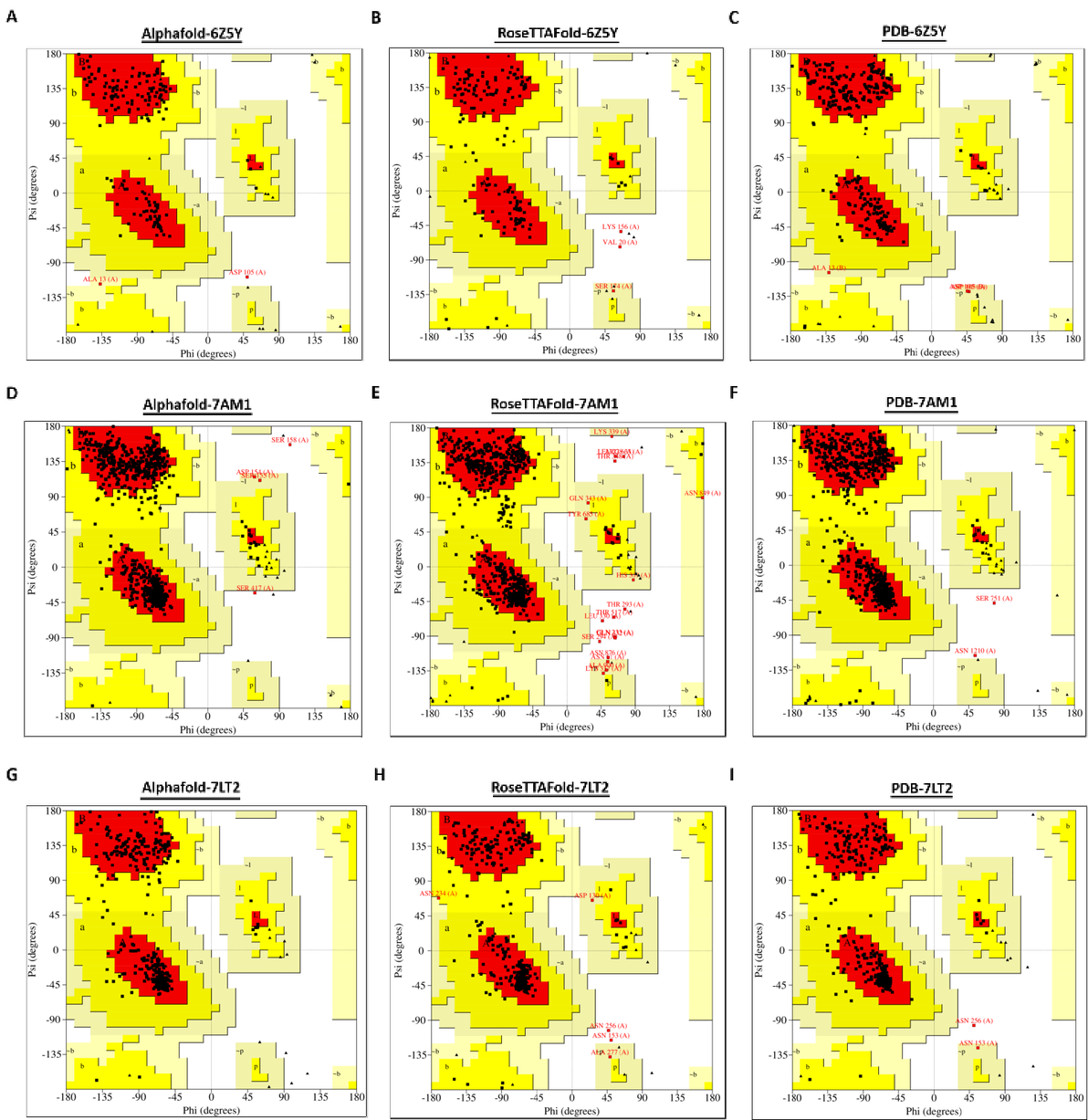
图 2. 蛋白6Z5Y,7AM1和7LT2的拉氏图
2.2 预测模型与对应晶体结构的相似度对比
蛋白质结构比对有很多方法,这里选择较为权威的RMSD、TM-score和Contact Map Overlap。
RMSD小于3Å时认为两个结构相似。TM-score衡量蛋白质结构之间的相似性而不依赖于蛋白质大小。TM-score大于0.5则认为结构为相似。最后通过Contact Map Overlap (CMO)观察结构在三维层面的差异,在本文中,如果蛋白质的两个残基主链碳原子的距离小于8Å,则判断它们接触。
2.2.1 已具有较高同源性蛋白结构的蛋白的预测模型与其对应晶体结构比较
首先这里将比较同源性较高的三个蛋白,7LA7,7M5I,7ONO的预测,这些蛋白在数据库中可以匹配到较多同源性模板,故而预测的准确性较高。
在7LA7,7M5I和7ONO蛋白的预测中Alphafold 和RoseTTAFold预测模型质量较好,二者预测的模型除了在末端一些无规则区域外其核心位置与晶体结构相似性高(图3.A,B)。7LA7,7M5I和7ONO的Alphafold模型的TM-score分别为0.96,0.98和0.98,RMSD为0.73Å,0.78Å和1.00Å;RoseTTAFold模型的TM-score为 0.93,0.9和0.94,RMSD为1.24Å,1.43Å和2.01Å;可见,在7LA7,7M5I和7ONO蛋白的预测中Alphafold 和RoseTTAFold预测模型与晶体结构的相似程度都较高。
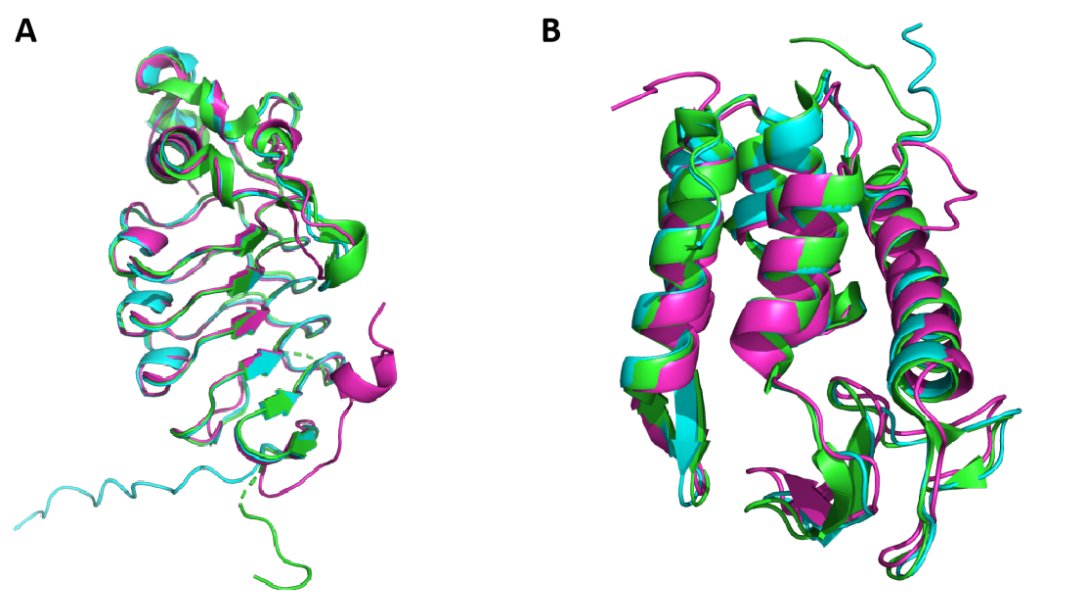
图 3. 7LA7,7M5I的Alphafold和RoseTTAFold模型与其晶体结构蛋白结构的重合图
为了进一步观察预测模型和蛋白晶体结构相似情况,我们使用了Contact map overlap (CMO),它可以将两个蛋白质的三维空间差异二维化,从而直观地比较蛋白结构的差别。7LA7,7M5I和7ONO的Alphafold模型的与其各自晶体结构的残基接触比例分别达到95.86%,95.23% 和94.56%。RoseTTAFold模型的与其各自晶体结构的接触比例分别达到87.23%,88.53%和93.34%。在7LA7,7M5I和7ONO中,Contact map overlap印证了之前RMSD和TM-score的结果,Alphafold 和RoseTTAFold的预测模型与晶体结构的相似程度较高,同时Alphafold模型与晶体结构更为相似。

图 4. 7LA7,7M5I和7ONO的Alphafold和RoseTTAFold模型与其晶体结构的Contact Map Overlap
2.2.2 Alphafold和RoseTTAFold对已具有低同源性蛋白结构的蛋白的预测模型分别与其对应晶体结构作比较
Alphafold 和RoseTTAFold在蛋白结构具有较高同源性蛋白的预测模型与其相应晶体结构的相似程度较高。而为了观察两个软件对低同源性蛋白结构预测的准确程度,这里选用6Z5Y,7AM1,7LT2三个蛋白结构同源性小于30%的蛋白。
Alphafold和RoseTTAFold的预测模型与6Z5Y晶体结构比较中,Alphafold模型与6Z5Y晶体结构的相似度比RoseTTAFold模型的高(图5.A),RoseTTAFold模型在α螺旋(图5.B,C),与晶体结构位置存在明显的偏差。6Z5Y晶体结构和Alphafold的模型比较的TM-score为 0.96;RMSD为0.91Å。RoseTTAFold的模型的TM-score为 0.86229,子RMSD为2.01 Å;在6Z5Y这个蛋白的预测上Alphafold 模型在与晶体结构的相似程度上较RoseTTAFold模型高。

图 5. 6Z5Y蛋白的Alphafold和RoseTTAFold模型与6Z5Y晶体结构蛋白结构的重合图
7AM1蛋白晶体结构81位谷氨酸(E)~150位天冬氨酸(N)的氨基酸残基缺失。将 Alphafold和RoseTTAFold模型与7AM1蛋白晶体结构重合(图6.A),可以明显可以观察到晶体结构蛋白缺失处,Alphafold和RoseTTAFold预测的结果大不相同,Alphafold给出的预测是一段自由摆动的无规则结构,RoseTTAFold给出的预测是几个α螺旋结构(图6.B)。与此同时RoseTTAFold的预测模型与晶体结构的相似度上存在较大的差异(图6.C)。由于晶体结构缺失区域电子密度差,根据经验蛋白质晶体结构中的缺失区域是那些无法解析的区域,7AM1的Alphafold模型的TM-score 为0.97,RMSD为1.41 Å;RoseTTAFold的模型得到的TM-score为 0.88,RMSD为3.38 Å;排除晶体结构的缺失区域,Alphafold 模型在与晶体结构的相似程度上较RoseTTAFold模型高。

图 6. 7AM1蛋白的Alphafold和RoseTTAFold模型与7AM1晶体结构蛋白结构的重合图
7LT2晶体结构和Alphafold预测模型的TM-score为 0.95;RMSD为1.74Å;7LT2晶体结构和RoseTTAFold预测模型的TM-score为 0.88,align的RMSD为3.01Å。在7LT2的预测中RoseTTAFold模型与晶体结构的相似性较6Z5Y,7AM1出现一定的差异其模型的RMSD甚至大于3Å。
在数据库中蛋白结构同源性小于30%的蛋白6Z5Y,7AM1和7LT2与其对应晶体结构的共同联系( Common contact)比同源性大于30%的蛋白的共同联系低,Alphafold的6Z5Y,7AM1和7LT2模型的共同联系率分别是98.69%, 97.07% 和97.03%。RoseTTAFold模型的为87.23%,88.53%和93.34%(图.8)。可见RoseTTAFold模型共同联率较Alphafold模型的低,Alphafold和RoseTTAFold模型的共同联系率也都比同源性大于30%的蛋白低。
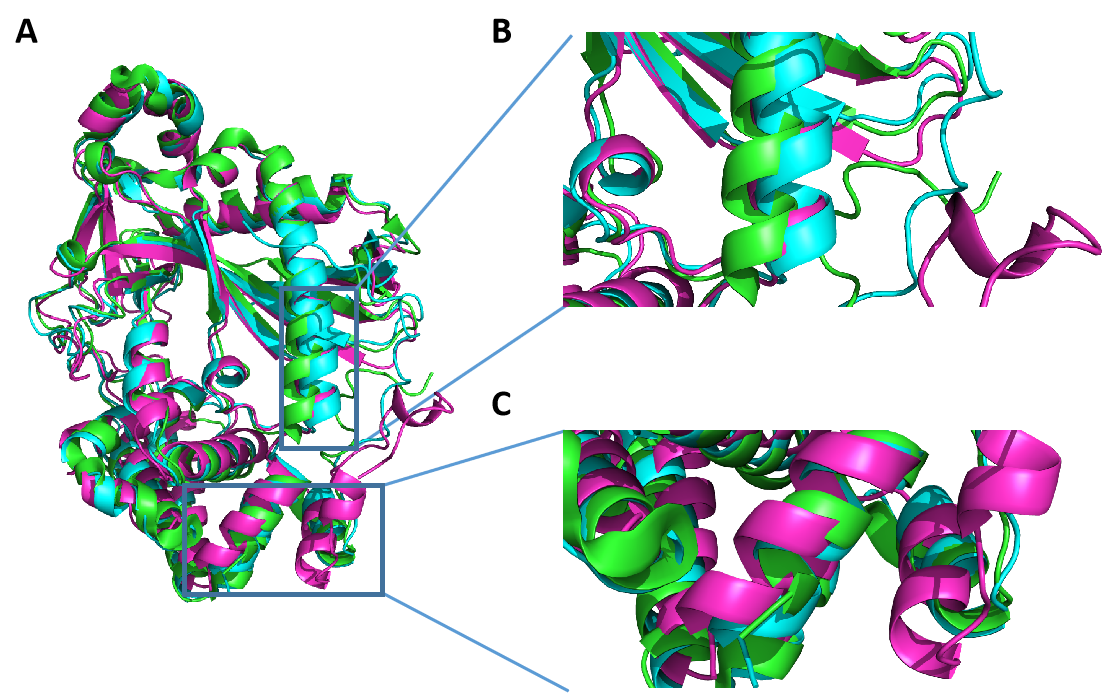
图7. 7 LT2蛋白的Alphafold和RoseTTAFold模型与 7 LT2晶体结构蛋白结构的重合图

图 8. 6Z5Y,7AM1和7LT2的Alphafold和RoseTTAFold模型与其晶体结构的Contact Map Overlap
总结与展望
Alphafold和RoseTTAFold在预测蛋白结构同源性不同的蛋白时,在蛋白结构同源性较高的蛋白中预测模型的质量较高,主要体现在拉氏图中位于最佳区域的氨基酸残基比例与晶体结构相接近,且位于拉氏图不允许区域的氨基酸残基比例较小甚至为零。相较之下蛋白结构同源性低于30%的蛋白的Alphafold和RoseTTAFold预测模型的质量会有所下降。
在预测模型与蛋白晶体结构的相似性比较方面,其具有较高蛋白结构同源性的蛋白的Alphafold和RoseTTAFold预测模型的质量和与晶体结构比较的相似性都比低同源性蛋白的模型要高,而且Alphafold模型的相似性都比RoseTTAFold模型的要高。(表1,表2)
Alphafold和RoseTTAFold作为人工智能在生命科学领域的应用代表,他们对人工智能在生物领域的应用有着至关重要的参考意义。
参考文献
[1] Branden, C.I. and J. Tooze, Introduction to protein structure. 2012: Garland Science.
[2] Brownlee, M., Biochemistry and molecular cell biology of diabetic complications. Nature, 2001. 414(6865): p. 813-820.
[3] Woolfson, M.M. and M.M. Woolfson, An introduction to X-ray crystallography. 1997: Cambridge University Press.
[4] Cavanagh, J., et al., Protein NMR spectroscopy: principles and practice. 1995: Elsevier.
[5] Ishima, R. and D.A. Torchia, Protein dynamics from NMR. Nature structural biology, 2000. 7(9): p. 740-743.
[6] Gonen, T., et al., Lipid–protein interactions in double-layered two-dimensional AQP0 crystals. Nature, 2005. 438(7068): p. 633-638.
[7] Liu, J., et al., Improving protein tertiary structure prediction by deep learning and distance prediction in CASP14. bioRxiv, 2021.
[8] Pereira, J., et al., High‐accuracy protein structure prediction in CASP14. Proteins: Structure, Function, and Bioinformatics, 2021.
[9] Jumper, J., et al., Highly accurate protein structure prediction with AlphaFold. Nature, 2021. 596(7873): p. 583-589.
[10] Baek, M., et al., Accurate prediction of protein structures and interactions using a three-track neural network. Science, 2021. 373(6557): p. 871-876.
[11] Pennisi, E., Protein structure prediction now easier, faster. 2021, American Association for the Advancement of Science.
[12] Bittrich, S., M. Schroeder, and D. Labudde, StructureDistiller: structural relevance scoring identifies the most informative entries of a contact map. Scientific reports, 2019. 9(1): p. 1-15.
[13] Joosten, R.P., et al., The PDB_REDO server for macromolecular structure model optimization. IUCrJ, 2014. 1(4): p. 213-220.
[14] Linge, J.P., et al., Refinement of protein structures in explicit solvent. Proteins: Structure, Function, and Bioinformatics, 2003. 50(3): p. 496-506.
[15] Coutsias, E.A., C. Seok, and K.A. Dill, Using quaternions to calculate RMSD. Journal of computational chemistry, 2004. 25(15): p. 1849-1857.
[16] Kirchmair, J., et al., Evaluation of the performance of 3D virtual screening protocols: RMSD comparisons, enrichment assessments, and decoy selection—what can we learn from earlier mistakes? Journal of computer-aided molecular design, 2008. 22(3): p. 213-228.
[17] Reva, B.A., A.V. Finkelstein, and J. Skolnick, What is the probability of a chance prediction of a protein structure with an rmsd of 6 å? Folding and Design, 1998. 3(2): p. 141-147.
[18] Dong, R., et al., mTM-align: an algorithm for fast and accurate multiple protein structure alignment. Bioinformatics, 2018. 34(10): p. 1719-1725.
[19] Zhang, Y. and J. Skolnick, TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic acids research, 2005. 33(7): p. 2302-2309.
[20] Xu, J. and Y. Zhang, How significant is a protein structure similarity with TM-score= 0.5? Bioinformatics, 2010. 26(7): p. 889-895.
[21] Bhattacharya, S. and D. Bhattacharya, Evaluating the significance of contact maps in low-homology protein modeling using contact-assisted threading. Scientific reports, 2020. 10(1): p. 1-13.
[22] Nordström, K.J., et al., Independent HHsearch, Needleman–Wunsch-based, and motif analyses reveal the overall hierarchy for most of the G protein-coupled receptor families. Molecular biology and evolution, 2011. 28(9): p. 2471-2480.
