

尔云间 一个专门做科研的团队
原创 小果 生信果
欢迎点赞+收藏+关注
今天小果就为大家带来宏基因组物种组成分析的两个软件Kraken2和Bracken的教程和结果解读。 使用这两个软件对数据进行物种注释和丰度估计之前,大家不要忘了用质控(fastp)和去宿主(bowtie2)的流程,不然注释出的结果会不准确。
Kraken21.安装Kraken2使用conda一键安装,(服务器必须要有anaconda或者miniconda):
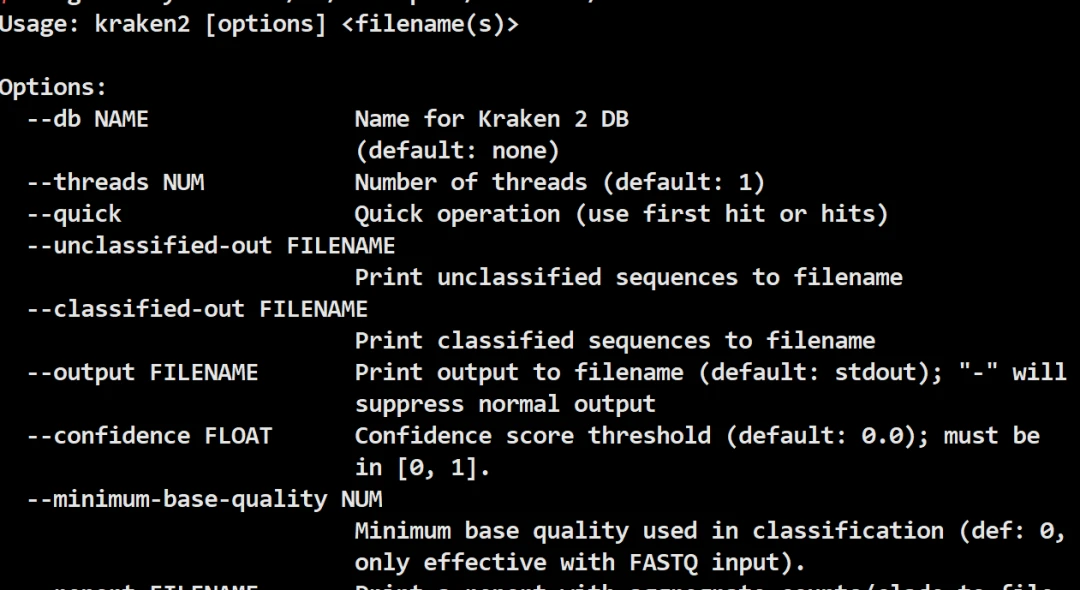
conda install -y kraken2输入kraken2 --help出现以下的提示就是按照成功:
2.数据库构建
kraken2 数据库地址:https://benlangmead.github.io/aws-indexes/k2,大家可以在上面下载自己需要的数据库到服务器上解压。

Kraken2数据库构建有三种方法方法1:直接下载官方提供的数据库(包括kraken2及bracken),解压即可
wget https://genome-idx.s3.amazonaws.com/kraken/k2_pluspf_20210517.tar.gz
wget https://genome-idx.s3.amazonaws.com/kraken/k2_pluspf_8gb_20210517.tar.gz
wget https://genome-idx.s3.amazonaws.com/kraken/k2_standard_20210517.tar.gz方法2:一步构建kraken标准数据库标准数据库:archaea, bacteria, viral, plasmid, human1, UniVec_Core,标准数据库比较大,有细菌,古菌,病毒等基因组数据,这是最简单的数据库构建方式,但是需要占用大量内存。
kraken2-build --standard --threads 4 --db path/to/standardDB
--standard是构建标准数据库的命令-
-db是数据库存放的路径
--threads 是线程数方法3:手动构建数据库可以从NCBI上下载序列进行建库,这个方法是最麻烦的,但是可以个性化进行建库,这个小果就不推荐大家使用啦。
## step1 从NCBI下载分类文件
kraken2-build \
--download-taxonomy \
--db ./K2db
## step2 从NCBI下载序列文件
kraken2-build \
--download-library fungi \ #下载真菌的数据库
--threads 4 \
--db ./K2db
## step3 建库
kraken2-build \
--build \
--threads 4 \
--db ./K2dbkraken2 建库完成后,假设我们把数据库放在db文件夹里面,那这里面就会有这三个文件:

3.运行 kraken2
kraken2 \
--threads 4 \
--quick \ # 快速模式
--paired \ # 输入数据为双端模式
--db path/to/db \ # 数据库路径
--report A1.kreport \ # 输出,report文件
--output A1.kraken \ # 输出,reads注释结果
path/to/A1_1.fq.gz \ # 输入,fq1
path/toA1_2.fq.gz # 输入,fq2会生成一个output文件和一个report文件,接下来小果为大家一一解读一下。 其中output文件总共有五列(如下图)。第一列:C/U,代表是否分类,C代表分类,U则代表未分类。第二列:测序数据read的 ID号 ,来自FASTA/FASTQ文件。第三列:分类ID号,(例:Mucilaginibacter ginsenosidivorax (分类id为862126);如果分类id号为0,说明没有分类)。第四列:测序read的长度。第五列:以空格分隔的列表,指示序列中每个k-mer的LCA映射 (冒号之前taxid,0为没有数据)。
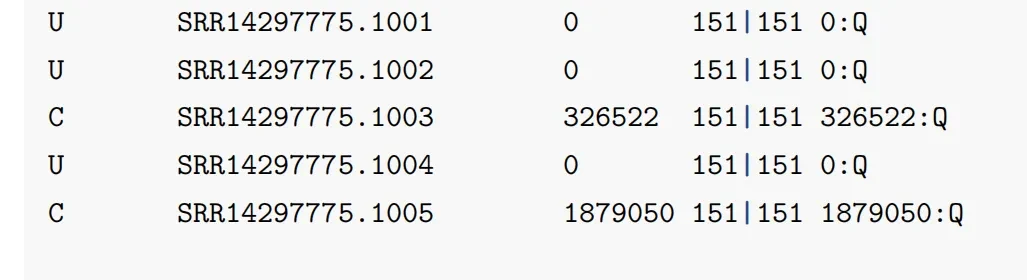
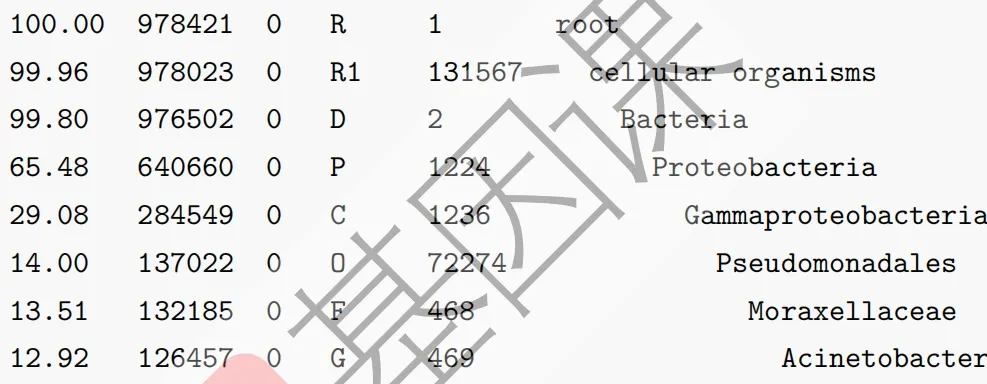
其中kreport文件总共有六列(如下图)。第一列:这个属或种的read在总read的比例。第二列:统计分类子集数量即该分类单元的read数,比如为属分类水平,会包括这个属之下的所有种的read数。第三列:只鉴定到这个分类单元的read数,比如属水平,就是只包含确定到这个属的read数,但不包括可以鉴定到这个属之下的种的read,这个是和第2列的区别。第四列:分类水平,(P)hylum(门), (C)lass(纲), (O)rder(目), (F)amily(科), (G)enus<属>, or (S)pecies<种>。第五列:NCBI分类号。第六列:缩进的该水平上的学名。

1.安装Bracken
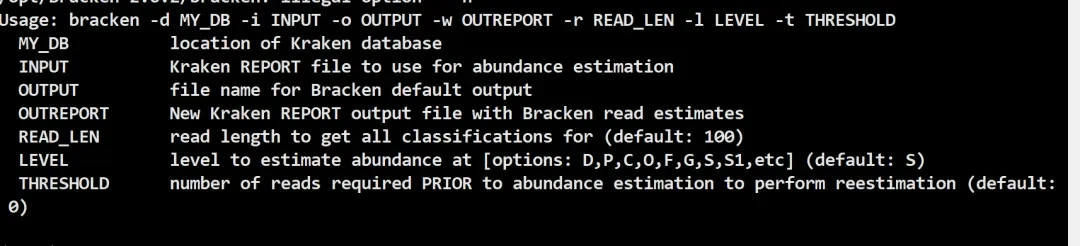
conda install -y bracken输入bracken -h出现以下信息就说明安装成功了。
2.Bracken建库
bracken-build \
-d path/to/db/ -t 4db是Kraken2数据库所放的文件夹,构建好Kraken2和Bracken的数据库之后,里面大概有如下图这些文件,大家可以作为参考。

3.运行brackenBracken可以进行多个水平的分析,这里为进行species水平分析,首先建立一个S文件夹,运行:
mkdir S
bracken \
-d path/to/db \ # 数据库路径
-i A1.kreport \ # 输入, 为kraken2 report文件
-o A1.bracken.S \ # 输出,物种丰度估计表格
-w A1.bracken.S.kreport \ # 输出,kraken2 report格式文件
-l S \ # 分类水平,如 D,P,C,O,F,G,S(界门纲目科属种)
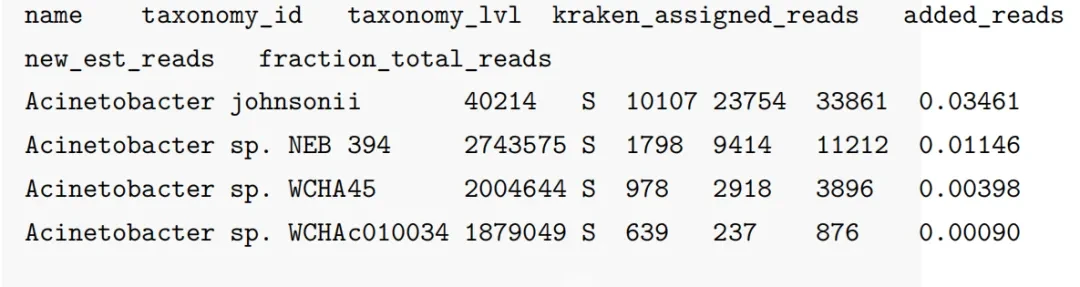
-t 4 # 线程数会生成一个output文件和一个report文件。output就是物种丰度估计表格,如下图所示,
report文件的格式则和kraken2中生成的一样,如下图。
好啦,这就是宏基因组物种组成分析Kraken2+Bracken的全部内容,这样我们就得到了每个样本的物种组成和丰度信息,接着就可以进行多样性分析了。希望大家可以继续跟着小果学习更多宏基因组方面的知识吧!
往期推荐
