
近年来,有机太阳电池的效率(OSCs)随小分子受体Y6及其衍生物的兴起而步入15%的时代,如图1所示,OSCs中给体分子和受体分子能级与电极功函数的匹配程度决定着OSCs对于载流子的收集能垒,对器件的光电性能至关重要[1],因此确定有机半导体的能级以实现能级的匹配非常重要。本文首先结合相关文献阐述了循环伏安法对小分子有机半导体能级的测量原理,接着论述了一些例如紫外光电子能谱(UPS)结合紫外-可见光谱(UV-vis)等也能够估算有机半导体能级的方法,最后结合文献讨论了一些在进行能级测量时经常遇到的误区。水平有限,如有不同看法非常欢迎讨论。
这里首先要说明的是,实验确定的-IP和-EA与HOMO和LUMO是概念上、数值上都完全不同两类事物,HOMO和LUMO理论计算的产物,是无法被测量的,但常有报道将这两者混为一谈,因此下文中所有对HOMO和LUMO的描述都是“估算”,这在本文的第三小节中有进一步阐述。
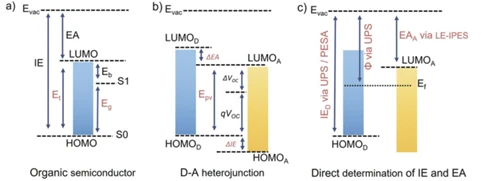
图1 有机太阳能电池的能级排列[1]
一、电化学循环伏安法测量有机半导体材料能级
循环伏安法(cyclic voltammetry, CV)是一种成本较低、门槛较低,操作简单,通过测量氧化还原电位来估计电离能IP(或HOMO)和电子亲合能EA(或 LUMO)的最广泛使用方法。
对于循环伏安法测量有机半导体能级的原理如图2所示,实验将有机半导体材料的电子电离能IP、电子亲合能EA和禁带宽度(Eg)与其起始氧化还原电位相关联。聚合物的起始氧化电位(E’ox)近似对应于IP;起始还原电位(E’red)对应于EA,起始氧化电位与起始还原电位之差则对应于聚合物的禁带宽度[2]。
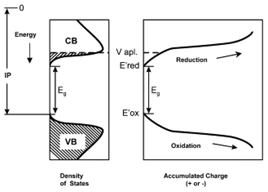
图 2从简化的聚合物能带结构推导出电化学测量的起始电位E’ox、E’red和IP、EA之间的关系[2]
循环伏安法的测量采用三电极体系,包括工作电极、对电极和参比电极。其基本原理是:相对于参比电极控制工作电极的电位在一定的电位范围内以三角波电位扫描(三角波电位扫描的速率一般为10 ~ 20 mV·s-1),与此同时自动测量并记录电位扫描过程中流过工作电极和对电极之间的电流。在对样品进行能隙测量时,往正电位方向扫描会观察到一对氧化和再还原的电流峰,在往负电位方向扫描时会观察到一对还原和再氧化的电流峰。通过对循环伏安曲线图正电位方向扫描的氧化电流曲线的起始部分做切线、负电位方向扫描的还原电流曲线的起始部分做切线,找出切线与横坐标(电位坐标)的交点即为材料的起始氧化电位E’red(图中记为φox)和起始还原电位E’ox(图中记为φred)[3],如图3所示。
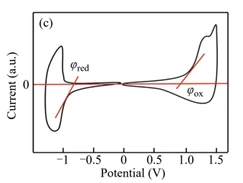
图3 有机半导体材料的循环伏安图[3]
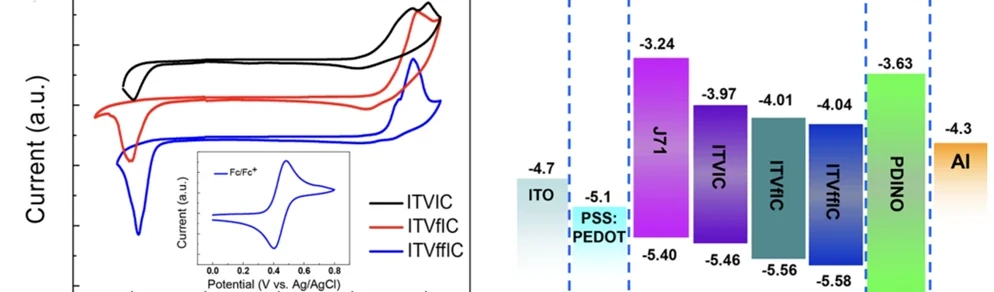
接下来以李永舫院士团队关于有机太阳能电池新型窄带隙受体的研究对分子能级的估算来举例。
该研究同样使用三电极体系,为了能测出薄膜状态下的材料性质,材料需要在电极上被制成固态薄膜而非配成稀溶液,对于薄膜的制备,一般采用将待测的有机半导体(共轭聚合物或共轭有机分子)溶于氯仿或甲苯等良溶剂中制成溶液,然后将它们的溶液滴涂到玻碳或铂工作电极的表面,随后晾干即可得到一层待测样品的薄膜。电解液使用0.1mol·L-1四丁基铵六氟磷酸盐(Bu4NPF6:易溶于极性溶剂的惰性电解质)的乙腈溶液。使用Ag/AgCl作为参比电极,由于其会受到电化学测量条件(比如不同的有机溶剂等)的影响,所以需要使用二茂铁氧化还原对(Fc+/Fc)作为内参比首先对参比电极进行标定(二茂铁在有机电解液中具有非常稳定和可逆的氧化还原特性)。常在计算能级时将二茂铁相对于真空能级的绝对能级定为 -4.8 eV。该报道中由于φ (Fc+/Fc) 在乙腈溶液中的值为0.09 V versus Ag+/Ag,使用关系式:
EHOMO/ELUMO≈-IP/-EA = −e(Eox/Ered + 4.71) (eV) (1.1)
可以计算出能级,结果如图4所示。
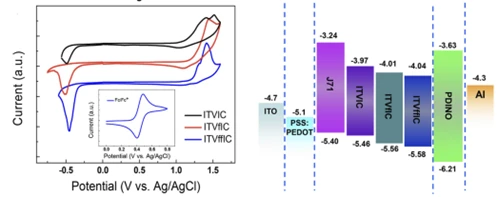
图 4 合成的有机半导体分子的循环伏安图及测得的材料能级及器件结构图[4]
二、其他获得有机半导体电子能级的方法
除了电化学循环伏安法之外,紫外光电子能谱法(UPS)是最常用的测量有机半导体分子电离能IP的办法。
UPS采用真空紫外线激发样品,其能量比较低(通常使用的光源为He I,波长为 58.4 nm,光子能量为 21.22 eV和 He Ⅱ,波长为30.4 nm,光子能量为40.8 eV,其中,He I光源具有单色性好、强度高等特点,是目前最常用的激发源)。光所激发的电子来自非常浅的样品表面(约1 nm),反映的是原子价层电子的信息,可以通过价层电子的能量分布获得有关价电子结构的各种信息,如价带谱、逸出功,以及价电子的分子电离势IP等,其中IP可以用来估算分子的HOMO能级。
其原理图如图5(a)[5]所示,材料逸出功Φ表示了从材料中移除一个电子所需的最小能量,在激发光源光子的能量(hv)已知的情况下,电子逸出功可以由激发光源光子的能量减去二次电子截止能量(Ecutoff)计算得到,最后将所得到的功函数与注入势垒(价带顶与费米能级之间能量差 ,此数值可以结合导电基底的费米能级获得)相结合就可以得到材料的电离能IP:
EHOMO ≈ -IP=-(Φ+EVBF )=-(hv-Ecutoff+EVBF) (2.1)
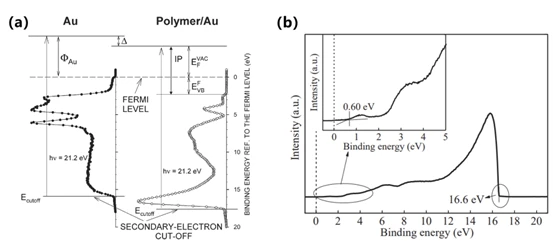
图 5 (a)UPS表征的表面和界面一些重要参数示意图[5];(b)实验所得的UPS数据图
以图5(b)所示,横坐标代表电子束缚能,纵坐标代表检测出来的光生电子经过信号放大后的相对强度。可以看到在12 eV之后谱线开始剧烈上升,表明有较强的二次非弹性散射电子出射。二次电子截止边对应被检测电子具有最高结合能的位置,即具有最低动能(可视作0)所对应的位置,通常结合费米边的位置用来确定材料的逸出功,即:
Φ= hv-Ecutoff (2.2)
通过截取可以得知二次电子截止能量Ecutoff为16.6 eV,当采用He I光源时,这个信号截断表明21.22 eV的光子能量最多只能激发结合能为16.6 eV的电子,使其不经过任何散射而到达样品表面,以式(2.2)得到材料的逸出功Φ为4.62 eV,同时考虑到相对于基底的注入势垒(EVBF)为0.60 eV,则该样品的IP应为:
IP = 21.22 - (16.6 - 0.60) = 4.62 + 0.60 = 5.22 eV (2.3)
与电化学循环伏安法测得的数据不同,电化学起始氧化/还原电位会受到有机半导体薄膜和金属电极之间存在的肖特基势垒的影响,而在UPS测量中不存在这一势垒,所以两者的数据可能会存在出入。
三、对能隙测量的一些误区
第一个需要注意的问题是,由于紫外光电子能谱(UPS)只能激发电子,因此使用UPS来测量能级时只能测得IP的值,当想确定材料的EA时,很多研究组直接使用通过UV-vis光谱所测出的光学能隙(optical gap)来间接计算,但是光学能隙和真实的能隙(fundamental gap)大小有很大的差别。
如下图所示[6],由于有机共轭材料中光激发的过程是从一个电子态(电子基态S0)到另一个电子态(电子激发态S1)的跃迁,即首先会形成激子态,电子和空穴具有激子结合能EB,因此它们的optical gap通常将明显低于fundamental gap;而从前后电性变化上来看,在CV测试中,从分子中抽取电子来测氧化势、向分子中注入电子来测还原势,即初始状态为中性,但最终状态总是带电的,此过程是存在克服静电排斥的,即库仑能量,而光学测量前后电性则是不变的,这都说明光学能隙和真实能隙从本质上是有区别的。
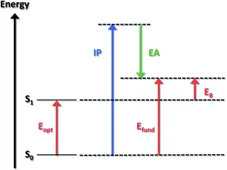
图 6 分子能隙的示意图[6]
还有一个相关文章经常出现的问题:将CV中测得的-IP和-EA认定为测得了HOMO和LUMO能级,而理论上来说,HOMO和LUMO是不可测得的。以下论述均引自卢天老师(开发Multiwfn多功能波函数分析程序,计算化学领域文章被引超过23000次)的博客[8]:
“从量子化学的角度来看,分子轨道是理论上做了单电子近似才有的概念,是将分子波函数以简化方式求解才诞生的,显然是纯粹人为虚构出来的产物。Hartree-Fock、半经验、KS-DFT方法全都可以得到分子轨道,具体来说,分子轨道是人为定义的单电子有效哈密顿算符的本征函数的统称。既然分子轨道在现实当中都不存在,谈何实验观测?在原理上怎么可能实验测定?显然,那些号称实验上测定的分子轨道根本就不是分子轨道,而是对观测到的数据的错误理解和解释,将其错误地冠上了分子轨道的名字。实际上,循环伏安法测的是溶液中的氧化势和还原势,这对应于电极反应过程的自由能变,离HOMO、LUMO能量隔着多达三层近似!显然把溶液中的氧化势和还原势直接分别当成HOMO和LUMO能量的负值是非常荒唐的,只能说存在正相关性而已,而这相关性还不怎么样。”
也就是说,CV测出的氧化势和还原势只能大致估算HOMO和LUMO(其只能由理论计算得到)。从理论计算的角度看,通过一种特定的理论计算路径和方法,比如在经过了忽略溶解自由能的近似、忽略自由能热校正量并近似为垂直过程以及Koopmans近似后,才能计算得到HOMO和LUMO能级。因此本文中所有论述实验的内容采用的都只使用了HOMO和LUMO的“近似测量”,“估算”的表达。
那么理论计算得到的HOMO和LUMO能否直接用于OSCs的器件评估呢?由于介电环境不同、固态环境下分子堆积导致的分子间相互作用,固态薄膜上测得的能隙将小于气相中的能隙。因此,为了能在OSCs中建立材料和性能关系,在实际情况下测量固体薄膜状态下的给体受体材料的IE和EA非常重要,而HOMO和LUMO的表述作为对孤立分子的理论计算结果,直接用做对于OSCs性能的分析凭证的说服力还是非常有限。
四、总结
本文的初始想法是通过几篇文献论述一下“循环伏安法对有机半导体HOMO和LUMO能级的测量”,但研究越深,越发现了不少值得深入的点,文章的题目也是一改再改。之前对于CV、UPS测量分子轨道能级的概念虽然有过怀疑——溶剂环境下的分子氧化势和还原势或是样品表面初最外层电子的电离能为什么就能和轨道能级划等号?但却没有深究其原理,在之前的文章写作中也直接采用了这样的方法,类似的还有将分子能隙的大小作为评判一个分子稳定性的标准,着实是想当然、缺失了原理上的思考,要吸取这样的经验,科学研究切忌人云亦云。
参考文献
[1] Bertrandie, J., Han, J., De, C. S. P., Yengel, E., Gorenflot, J., Anthopoulos, T., Laquai, F., Sharma, A., Baran, D., The Energy Level Conundrum of Organic Semiconductors in Solar Cells. Adv. Mater. 2022, 34, 2202575.
[2] Micaroni L, Nart F, Hümmelgen I. Considerations about the electrochemical estimation of the ionization potential of conducting polymers[J]. Journal of Solid State Electrochemistry, 2002, 7: 55-59.
[3] Li X, Li Y. Measurement of Electronic Energy Levels of Conjugated Polymers and Organic Molecules[J]. Acta Polymerica Sinica, 2022, 53(8): 995-1004.
[4] Li X, Huang H, Bin H, et al. Synthesis and photovoltaic properties of a series of narrow bandgap organic semiconductor acceptors with their absorption edge reaching 900 nm[J]. Chemistry of Materials, 2017, 29(23): 10130-10138.
[5] Braun, S., Salaneck, W.R. and Fahlman, M. (2009), Energy-Level Alignment at Organic/Metal and Organic/Organic Interfaces. Adv. Mater., 21: 1450-1472.
[6] Bredas J L. Mind the gap![J]. Materials Horizons, 2014, 1(1): 17-19.
[7] Sharifzadeh S, Biller A, Kronik L, et al. Quasiparticle and optical spectroscopy of the organic semiconductors pentacene and PTCDA from first principles[J]. Physical Review B, 2012, 85(12): 125307.
[8] 正确地认识分子的能隙(gap)、HOMO和LUMO, http://sobereva.com/543.
