
ATAC-seq分析干货
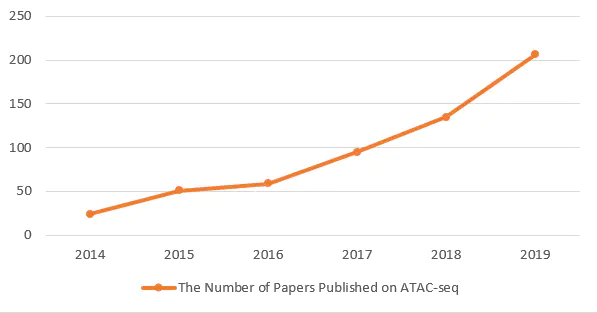
ATAC-seq技术由于其要求细胞量少,实验简单、快速、高效且应用范围广,是近年来转录调控、表观遗传修饰研究的一项重要的技术手段。近两年应用ATAC-seq方法发表的文章数量也是飞速上升,可见ATAC-seq的火爆程度。现在,如果你还不了解ATAC,还不抓紧学起来!今天我们先来学习一下ATAC-seq相关的背景介绍。
2014年至2019年ATAC-seq技术相关文章统计
背景介绍
01.染色质可及性介绍
在介绍ATAC-seq之前,不得不提的就是染色质可及性的概念。大家知道,真核生物的DNA会与组蛋白结合形成核小体,核小体进一步压缩折叠形成具有高级结构的染色体,染色体的高级结构在细胞周期的不同的时期压缩折叠程度不同,间期比较松散,此时的状态被称为染色质,用于和中期高度压缩的染色体进行区分。
研究者发现DNA内切酶可以对染色质进行切割,这些切割位点称为DNA超敏感位点。DNA超敏感位点位于没有组蛋白包裹的DNA片段上,这些位点的分布往往具有一定的规律性——切割后的DNA片段都在100-200bp左右。这些可以被切割下来的裸露的100-200bp的DNA片段称为开放染色质。后期进一步的研究发现,开放染色质通常是转录因子、增强子、绝缘子或者其他调控蛋白结合的片段,结合的过程仿佛是触发了细胞内的开关,可以影响细胞内基因复制以及调控基因的转录活性。DNA的这种被结合的特性称为染色质的可及性(chromatin accessibility)。
染色质的可及性是近些年的研究热点。由于染色质的开放程度是动态变化的,这种变化是影响基因表达调控的关键决定因素,在细胞分化、发育以及各类疾病的发生发展研究中具有重要的作用。
02.染色质可及性技术
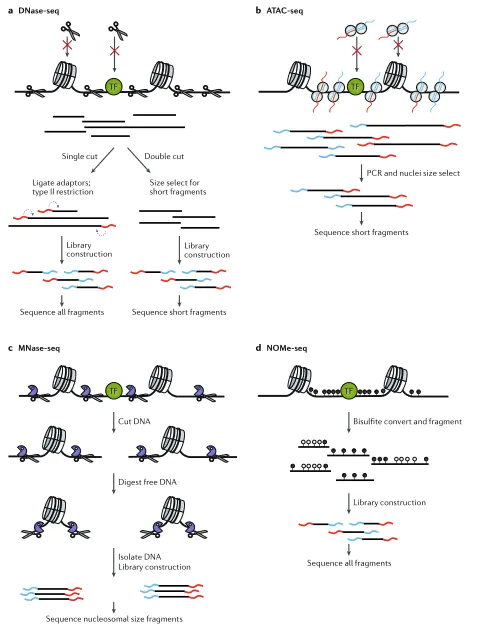
目前研究染色质可及性的方法总结主要有以下几种技术:MNase-seq、DNase-seq、FAIRE-seq、NOMe-seq以及ChIP-seq和ATAC-seq技术。
MNase-seq[1]是使用内切核糖酶--微球菌核酸酶(micrococcal nuclease, MNase)处理染色质,由于MNase同时具备核酸外切酶和内切酶活性,所以在对DNA进行消化时,会将裸露的DNA全部切割掉,直到遇到核小体或DNA结合蛋白等阻遏物保护,因此MNase-seq技术是将含有核小体包裹的区域切割下来进行测序,然后通过反向比较获得开放染色质区域。
DNase-seq[2]与MNase-seq技术互补,利用脱氧核糖核酸酶(DNaseI)识别DNaseI敏感位点,将裸露的DNA片段切割下来进行测序。
FAIRE-seq[3]技术是利用甲醛将染色质中裸露的DNA进行固定,随后进行超声波打断,再利用传统的DNA提取方法——酚氯仿抽提,来分离打断的DNA,从而进行高通量测序。
NOMe-seq[4]技术是通过人工甲基化修饰的方法进行核小体定位。使用GpC甲基转移酶处理固定的染色质,使未被核小体以及其他结合蛋白保护的GpC二核苷发生甲基化,由于基因组中含有大量的GpC,通过酶的处理可对核小体足迹进行高分辨的定位。
ChIP-seq[5]技术是通过染色质免疫共沉淀及时特异性地富集目的蛋白结合的DNA片段进行高通量测序,常用于特定转录因子或组蛋白特异性修饰位点的研究。
ATAC-seq[6]技术通过转座酶Tn5容易结合在开放染色质(未经蛋白或核小体保护的DNA部位)的特性,在整个基因组范围内对Tn5酶捕获到的DNA片段进行高通量测序。
染色质可及性研究技术概览
ATAC-seq技术介绍
1.技术原理
ATAC-seq(Assay for Transposase-Accessible Chromatin with highthroughput sequencing),利用超活性的Tn5转座酶容易结合在开放染色质的特性,通过Tn5酶切割基因组,在对切割下来的序列形成的转座酶复合物上,连接测序的barcode,构建成测序文库,并对测序文库进行测序,获得结合区域相关信息。
ATAC反应原理示2.优势
建库简单快捷,耗时短,重复性好
需要的细胞数目较少(一般500-50000个)
全基因组范围内检测
同时检测开放的DNA区域和被核小体占据的区域
3.ATAC-seq实验的几个建议
良好的实验设计以及操作是ATAC-seq成功的关键,关于实验策略有如下建议[8]:
生物学重复:最好有两个生物学重复,而且2个重复足够了
对照:最好使用对照,可以用不使用转座酶Tn5处理的样本做对照
PCR扩增:扩增次数尽量少,以减少PCR引入dup
数据量:测序数据量一般跟基因组大小有关,对于人类样本,推荐50M/样本。一般来说测序数据越多,比对结果会越准确,尤其对于复杂基因组,如:高重复基因组,可以提升比对准确性
测序策略:推荐使用paired-end,原因有以下几个方面:a.能够更准确的鉴定转座酶的插入位置,确定插入片段长度。单端测序虽然可以通过计算模型去推断片段长度,但是没有那么准确。b.双端测序对于比对软件,更容易更准确的鉴定dup的reads,提高分析结果的准确性。
分析步骤
1.原始下机数据的过滤质控
背景中介绍到,用来结合转录因子的染色质开放区域一般为100-200bp的小片段。随着测序技术的发展,我们ATAC-seq进行的高通量测序的策略一般是PE150(之前大部分为SE50),因此双端测序能测的插入片段大约在350bp左右。在这种情况下,我们实际测出的reads会把Tn5酶捕获到的片段测通,为了最大程度的利用测序数据,我们在数据清洗过程中一般会用Trim的形式进行过滤,即将reads中判断为接头的部分截取掉。
过滤的主要目的:1.去除接头污染;2.去除低质量reads
使用Trimmomatic软件进行过滤
过滤完成,得到clean数据后,就可以进行数据的比对、质控,以及各种可视化展示和peak calling分析了。后续分析内容,我们下期继续进行分享,感兴趣的小伙伴,赶快关注我们吧~
分析步骤
前期我们通过Trimmomatic软件对原始下机数据进行了数据清洗,主要包括:
去除下机数据中的接头污染
去除低质量的reads
从而获得clean reads数据。那接下来就可以将clean数据跟参考基因组进行比对,进行后续的可视化以及peak calling相关分析了。
1.数据比对(Bowtie2,picard-tools)
1.1 比对
1.2 过滤(去dup,去线粒体,低质量,未比对上的序列) 比对完之后,需要对比对得到的bam文件进行过滤处理。主要考虑过滤以下几个方面:
去除PCR duplicate reads,因为影响后续call peak
线粒体DNA和叶绿体DNA序列。由于线粒体和叶绿体DNA是裸露的,可以被Tn5酶识别切割,因此数据中会有线粒体和叶绿体的污染,比对完之后需要去掉(如果是植物,还需要去掉叶绿体)
低质量以及未比对上的reads
1.3 插入片段长度统计(picard-tools),同时生成call peak的bed文件) 插入片段长度是重要的评估实验好坏的指标,统计出的插入片段长度应该符合实验预期的长度。可以用picard-tools工具的CollectInsertSizeMetrics功能进行统计。
为了更方便的进行peak calling,可以根据插入片段起始终止位置整理成bed文件作为输入(需要注意的是bed文件的染色体起始位置是0起始的,第一个碱基的位置标记为0,而终止位置是从1开始的)。
根据ATAC-seq的实验原理,call peak 时,可以进行Tn5边界调整,将bed文件上下游各移动75bp,从而直接进行calling,而不用再进行reads延伸[1],具体call peak操作可以往下看。
2.比对结果可视化(deeptools,samtools)
得到比对结果之后,我们就可以图形化的展示peak的富集情况,peak reads在染色体的分布情况等等。另外,通常bam文件是比较大的,所以为了更加方便的使用IGV可视化的展示reads情况,通常会把bam文件转为bigwig文件形式。 2.1 bam转bigwig文件(deeptools,bamCoverage) bam的格式转换可以使用deeptools的bamCoverage工具,具体使用代码参考如下:
2.2 覆盖度(samtools) 使用samtools的mpileup工具统计base\reads在各个染色体的覆盖度情况,并进行可视化展示。
3.TSS、TES区域可视化
计算TSS或者TES附近的信号强度,可以使用deeptools[2]的computeMatrix工具进行。computeMatrix具有两个模式:scale-region和reference-point。
scale-region用来计算某一个区域内信号分布,比如如下代码设置的TSS和TES区域以及上下游各2K的区域内的信号分布计算;
reference-point可以计算相对于某一个点的信号分布情况。
computeMatrix工具计算信号分布情况结果,可以使用plotProfile以折线图的方式对覆盖区域信号分布进行可视化,也可以使用plotHeatmap以热图的方式对覆盖区域信号分布进行可视化。
TSS\TES区域可视化结果示例图
4.链互相关性SCC分析(phantompeakqualtools)
SCC(Strand Cross Correlation analysis)是可用于评估ATAC/Chip实验质量好坏的一个重要指标(转录因子富集),其原理基于peak的reads富集分布规律[3]。
第一:peak位点附近的正负链上reads分布相同;
第二:reads分布的中心点和peak的中心点存在偏移,如果将reads的位置移动一定的距离之后,正负链的中心重合,上下成对称分布。
用泊松相关系数来分析正负链测序深度的相关性,当正负链的中心点重合时,相关系数最高,可以有效衡量偏移过程。由此,可以得到偏移距离和相关系数之间的对应关系。因此,一方面,计算SCC有助于了解正负链间的相关性系数,另一方面可以检测Reads富集的程度来富集实验是否成功。
Chip-seq数据检测蛋白结合位点
典型的链交叉相关图会产生两个峰:
一个富集峰与主要的插入片段长度相关(高相关性),为peak峰。
另一个与测序read长度相关,被称为"phantom "peak。
一个高质量的转录因子富集数据,peak对应的峰最高,phantom peak对应的峰较矮, 如果两种峰都能够观测到,而phantom peak最高,则说明抗体还是富集到了部分序列,但是背景噪声太高了,不利于后续分析;而如果观测不到 peak峰,则说明富集实验是失败的。
Examples of Cross-Correlation Plots
为了更精准的进行判断,研究者提出两个量化指标:
Normalized strand cross-correlation coefficent (NSC)是最大交叉相关值除以背景交叉相关的比率,NSC值越大表明富集效果越好,NSC值低于1.1,表明较弱的富集,小于1表示无富集。NSC值稍微低于1.05,表明有较低的信噪比或很少的峰;
Relative strand cross-correlation coefficient (RSC),片段长度相关值减去背景相关值除以phantom-peak相关值减去背景相关值。RSC的最小值可能是0,表示无信号;富集好的实验RSC值大于1;低于1表示质量低。
5.Peak Calling
Model-based Analysis of ChIP-Seq (MACS2) 是用来检测DNA片段富集的软件,尽管当时是针对Chip-seq开发的,但是其非常好的适用于ATAC-seq。现在ATAC-seq主流的call peak软件依然是MACS2。
蛋白或转录因子结合位点附近会有reads富集,MACS2根据一定的统计模型,扫描全基因组,构建双峰模型,结合对照(background)来检测富集片段中真正的结合位点。MACS2可以鉴定Chip-seq中两种主要类型的富集模式:
组蛋白修饰的broad peaks或者broad domains
转录因子结合的narrow peaks。
而对于ATAC-seq而言,捕获的是染色质开放区域,可类似于Chip-seq中转录因子的检测模式。
MACS2 Peak Calling原理
由于ATAC-seq检测固定的染色质开放区域,所以MACS2检测ATAC peak时,可以使用--nomode1参数,不需要MACS去构建模型,同时需要设置--extsize参数和--shift参数。
--extsize参数和--shift参数是非常重要可以明显左右检测结果的参数,可以根据实验方法以及目的,设置相应的值。具体的脚本可以参考C. elegans的ATAC-seq分析[1]或者文档ATACSeq Pipeline,相关的参数和结果文件说明可以参考MACS2作者的github,MACS2。
考虑到ATAC-seq是通过Tn5酶结合的染色质开放区域,传统的使用MACS2软件(使用--extsize,--shift参数)检测ATAC peak的方式可能并不是最优的,比对上的成对的reads可能会被丢弃,从而忽略很多有效的序列信息。基于此,Harvard John M. Gaspar团队[5]推荐使用Genrich(https://github.com/jsh58/Genrich)软件,Genrich有特定的针对ATAC的分析模式,以Tn5转座酶切割位点为中心进行拓展,而不是通过拓展reads来检测peak。Genrich对线粒体reads、PCR dup reads、多重比对reads、生物学重复都有较好的处理,感兴趣的可以尝试使用此软件分析。
6.FRiP的统计
检峰之后,可以对检测到的peak数进行基本的个数、长度等统计。FRiP(Fraction of reads in peaks)[6],peaks中的reads与总reads的比例,即文库中结合位点片段占背景reads的比例,可理解为“信噪比”,也是样本富集效果的评价指标,可一定程度上反应富集效果。
通常,一个典型质量好的TF富集FRiP值约5%或者更高,大部分(787 of 1052)ENCODE数据集中FRiP值在1%以上,但低于此阈值的并不能说明富集实验是失败的,如ZNF274以及人类RNA聚合酶III有非常少的结合位点,所以具体需要综合实验以及研究的对象结合FRiP进行评估。
经过以上分析后,ATAC-seq分析的主要结果就已经有了,后续可以基于分析得到的peak结合自己的实验课题进行各种可视化展示、peak注释、差异分析、功能富集以及Motif分析等等,筛选出跟课题相关的结合位点。现在,感兴趣的小伙伴就可以上手啦~
